

Functional screening of lysosomal storage disorder genes identifies modifiers of alpha-synuclein neurotoxicity
- PMID: 37200393
- PMCID: PMC10231792
- DOI: 10.1371/journal.pgen.1010760
Functional screening of lysosomal storage disorder genes identifies modifiers of alpha-synuclein neurotoxicity
Abstract
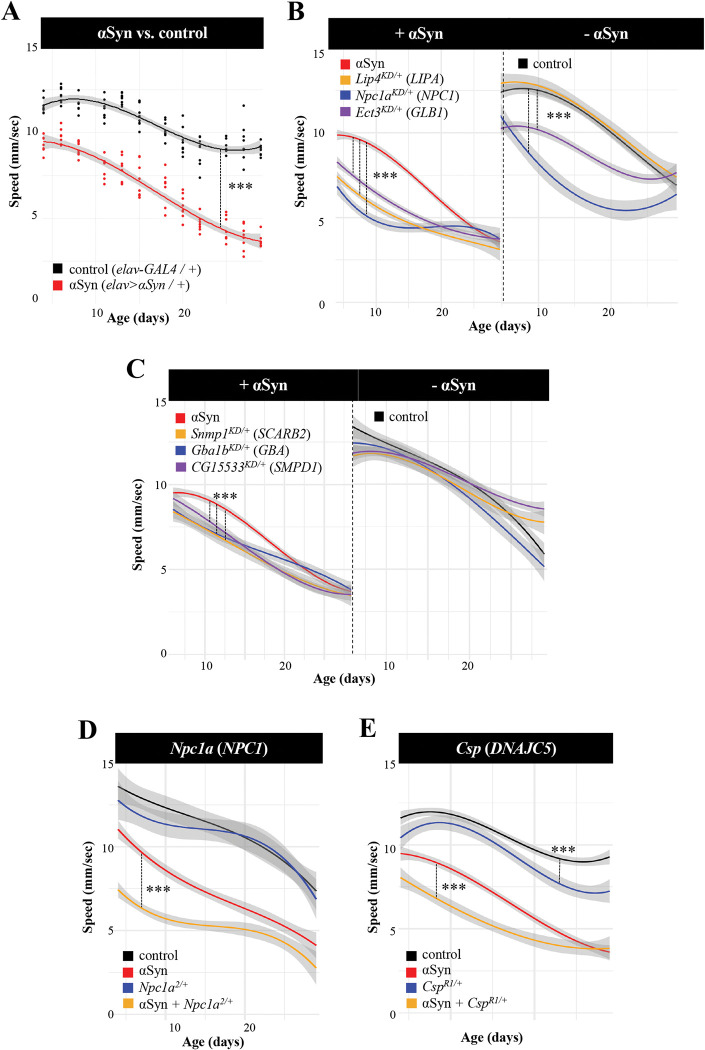
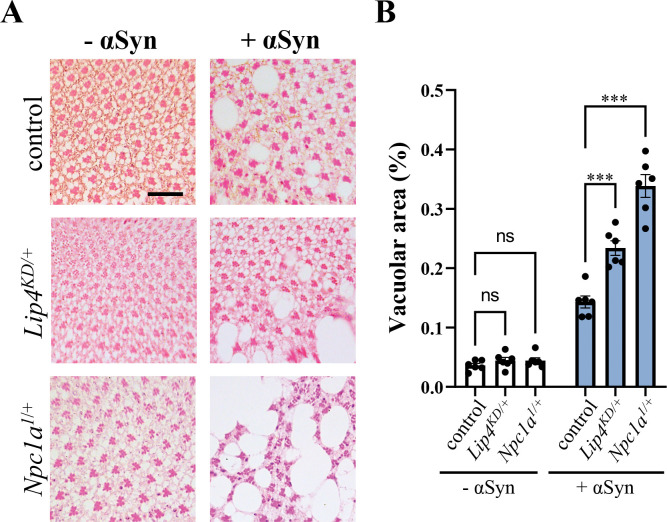
Heterozygous variants in the glucocerebrosidase (GBA) gene are common and potent risk factors for Parkinson's disease (PD). GBA also causes the autosomal recessive lysosomal storage disorder (LSD), Gaucher disease, and emerging evidence from human genetics implicates many other LSD genes in PD susceptibility. We have systemically tested 86 conserved fly homologs of 37 human LSD genes for requirements in the aging adult Drosophila brain and for potential genetic interactions with neurodegeneration caused by α-synuclein (αSyn), which forms Lewy body pathology in PD. Our screen identifies 15 genetic enhancers of αSyn-induced progressive locomotor dysfunction, including knockdown of fly homologs of GBA and other LSD genes with independent support as PD susceptibility factors from human genetics (SCARB2, SMPD1, CTSD, GNPTAB, SLC17A5). For several genes, results from multiple alleles suggest dose-sensitivity and context-dependent pleiotropy in the presence or absence of αSyn. Homologs of two genes causing cholesterol storage disorders, Npc1a / NPC1 and Lip4 / LIPA, were independently confirmed as loss-of-function enhancers of αSyn-induced retinal degeneration. The enzymes encoded by several modifier genes are upregulated in αSyn transgenic flies, based on unbiased proteomics, revealing a possible, albeit ineffective, compensatory response. Overall, our results reinforce the important role of lysosomal genes in brain health and PD pathogenesis, and implicate several metabolic pathways, including cholesterol homeostasis, in αSyn-mediated neurotoxicity.
Copyright: © 2023 Yu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Pastores GM, Hughes DA. Gaucher Disease. 2018. Jun 21; Available from: http://europepmc.org/books/NBK1269
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
