

Butyrate limits inflammatory macrophage niche in NASH
- PMID: 37202387
- PMCID: PMC10195803
- DOI: 10.1038/s41419-023-05853-6
Butyrate limits inflammatory macrophage niche in NASH
Abstract
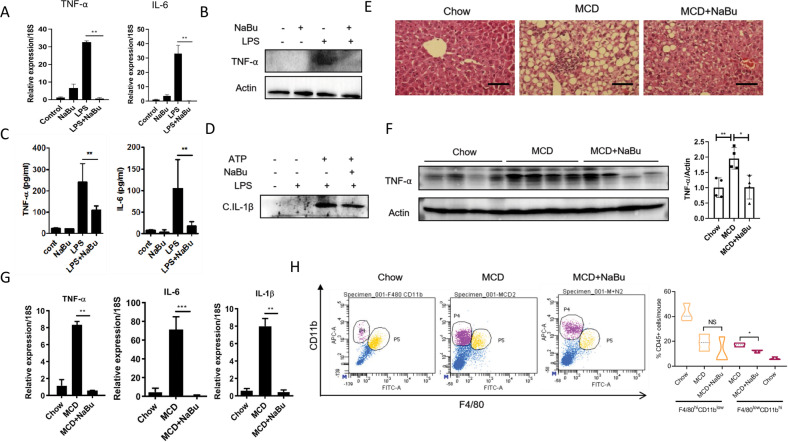
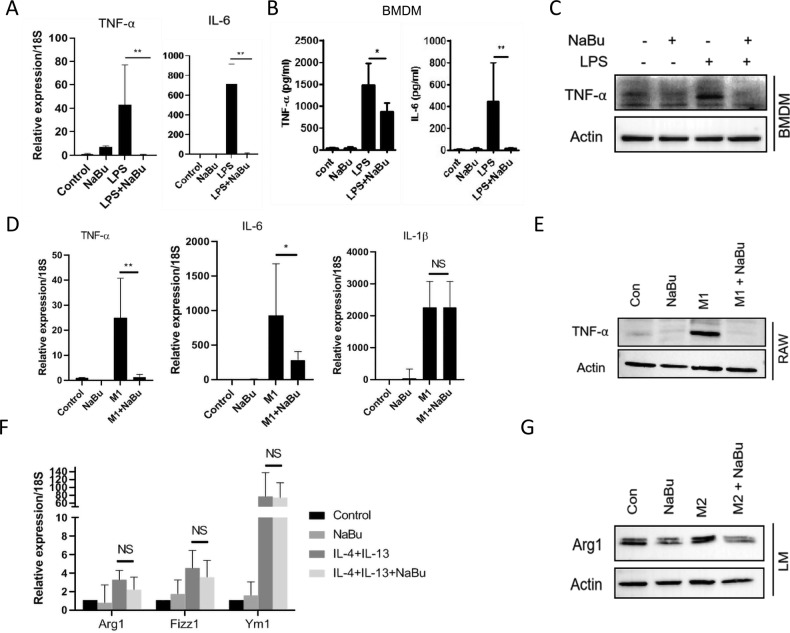
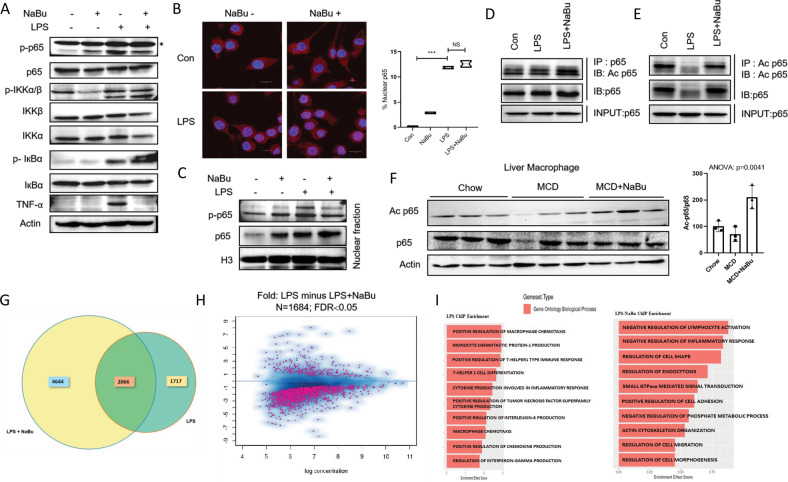
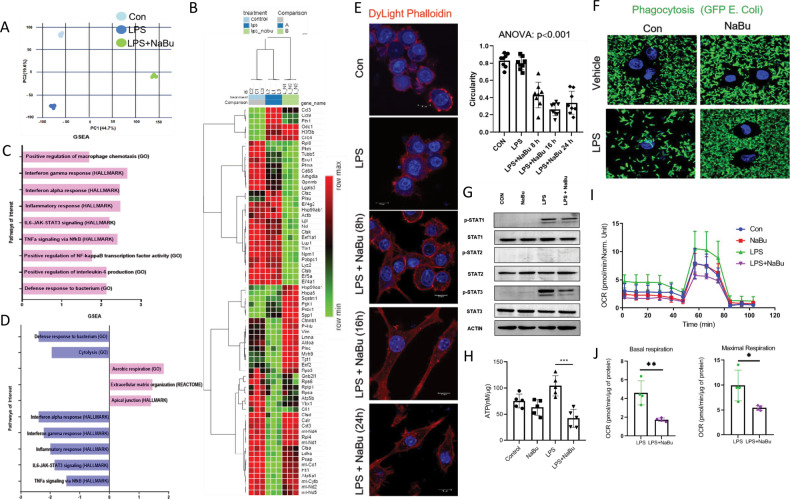
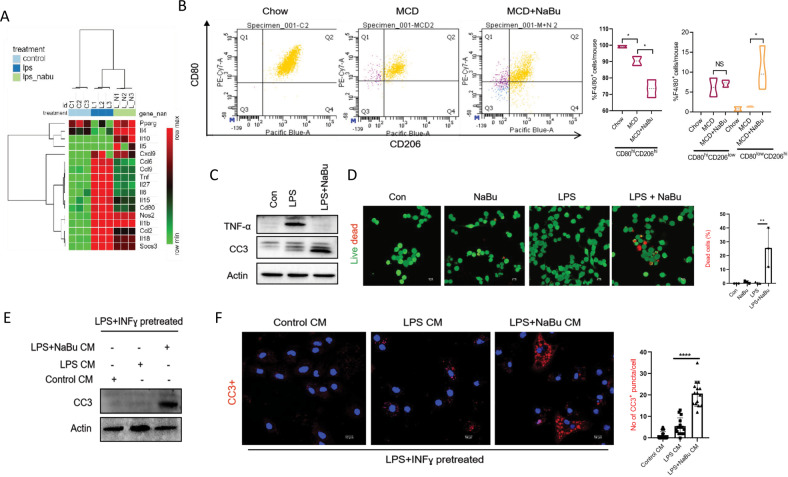
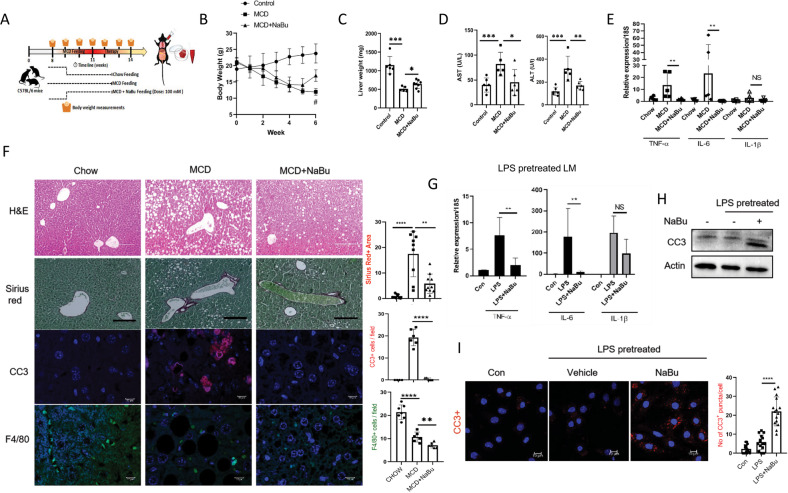
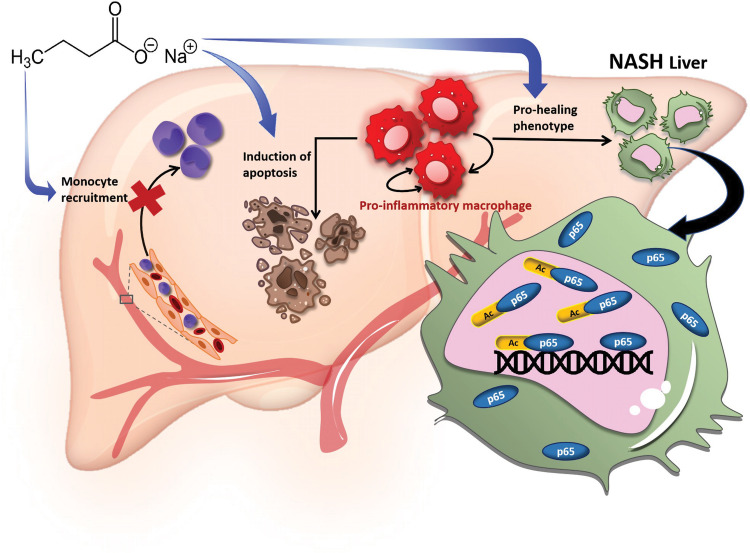
Immune cell infiltrations with lobular inflammation in the background of steatosis and deregulated gut-liver axis are the cardinal features of non-alcoholic steatohepatitis (NASH). An array of gut microbiota-derived metabolites including short-chain fatty acids (SCFA) multifariously modulates NASH pathogenesis. However, the molecular basis for the favorable impact of sodium butyrate (NaBu), a gut microbiota-derived SCFA, on the immunometabolic homeostasis in NASH remains elusive. We show that NaBu imparts a robust anti-inflammatory effect in lipopolysaccharide (LPS) stimulated or classically activated M1 polarized macrophages and in the diet-induced murine NASH model. Moreover, it impedes monocyte-derived inflammatory macrophage recruitment in liver parenchyma and induces apoptosis of proinflammatory liver macrophages (LM) in NASH livers. Mechanistically, by histone deactylase (HDAC) inhibition NaBu enhanced acetylation of canonical NF-κB subunit p65 along with its differential recruitment to the proinflammatory gene promoters independent of its nuclear translocation. NaBu-treated macrophages thus exhibit transcriptomic signatures that corroborate with a M2-like prohealing phenotype. NaBu quelled LPS-mediated catabolism and phagocytosis of macrophages, exhibited a differential secretome which consequently resulted in skewing toward prohealing phenotype and induced death of proinflammatory macrophages to abrogate metaflammation in vitro and in vivo. Thus NaBu could be a potential therapeutic as well as preventive agent in mitigating NASH.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
