

RNA stability controlled by m6A methylation contributes to X-to-autosome dosage compensation in mammals
- PMID: 37202476
- PMCID: PMC10442230
- DOI: 10.1038/s41594-023-00997-7
RNA stability controlled by m6A methylation contributes to X-to-autosome dosage compensation in mammals
Erratum in
-
Author Correction: RNA stability controlled by m6A methylation contributes to X-to-autosome dosage compensation in mammals.Nat Struct Mol Biol. 2023 Oct;30(10):1607. doi: 10.1038/s41594-023-01053-0. Nat Struct Mol Biol. 2023. PMID: 37415011 Free PMC article. No abstract available.
Abstract
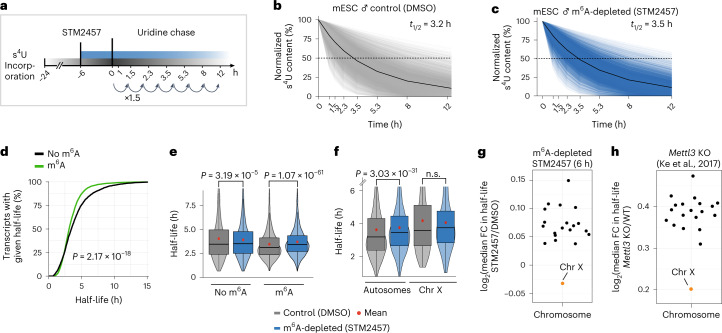
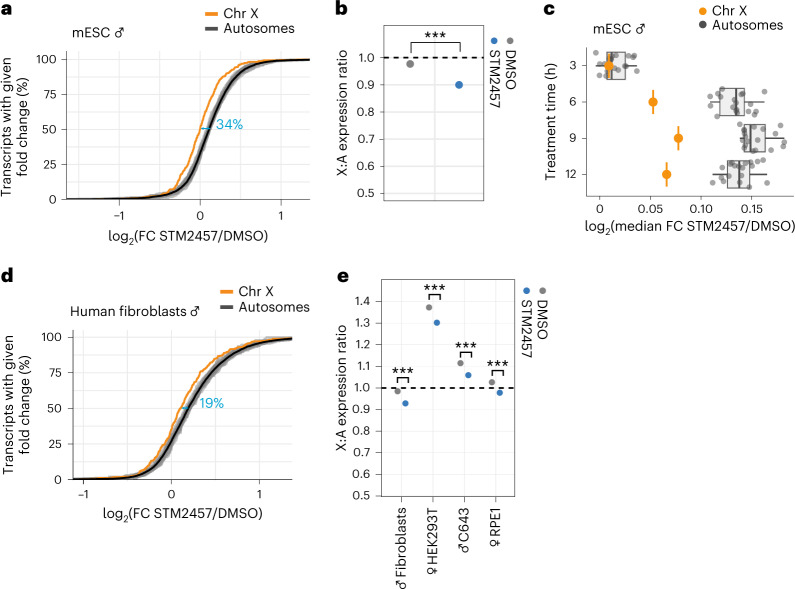
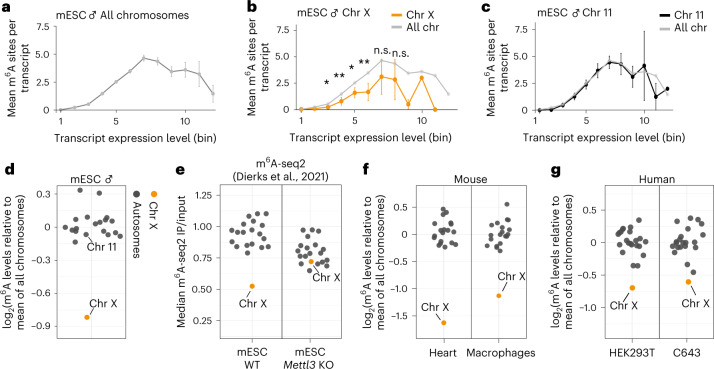
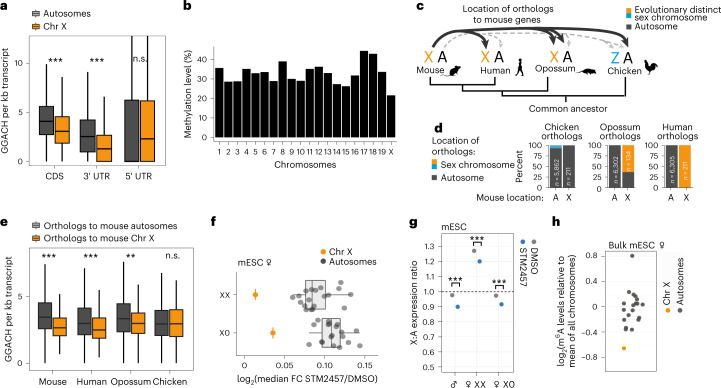
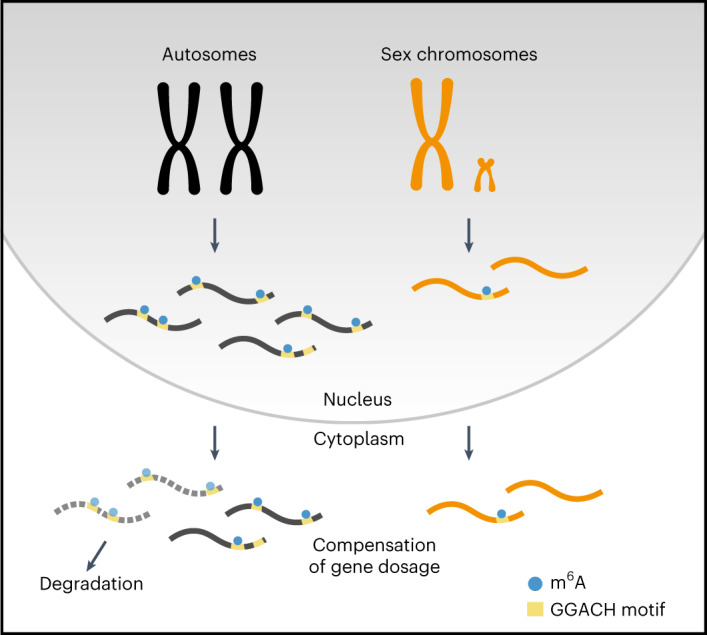
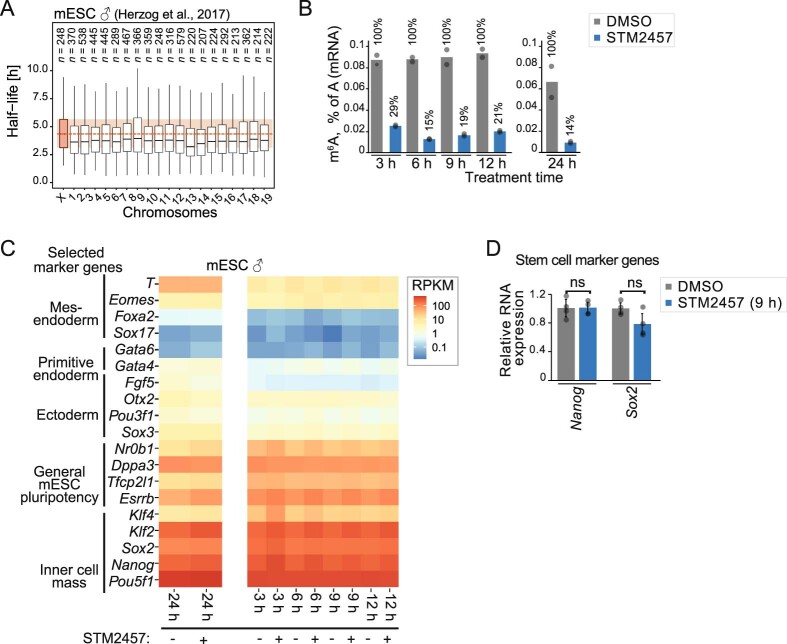
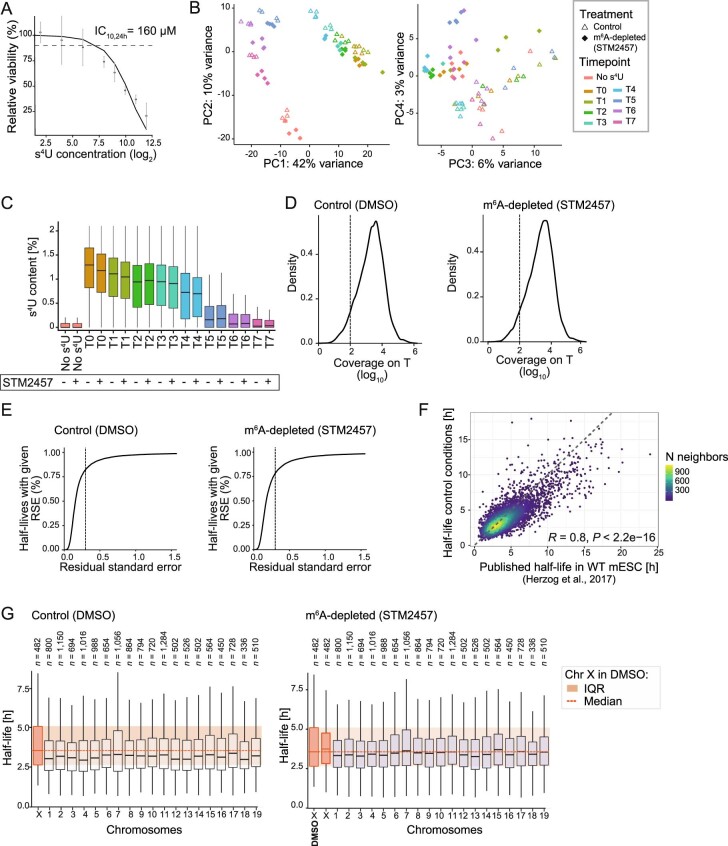
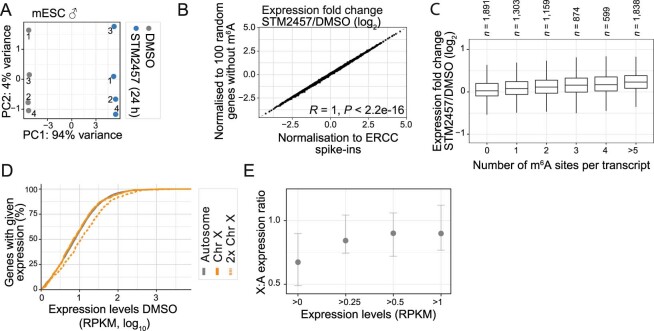
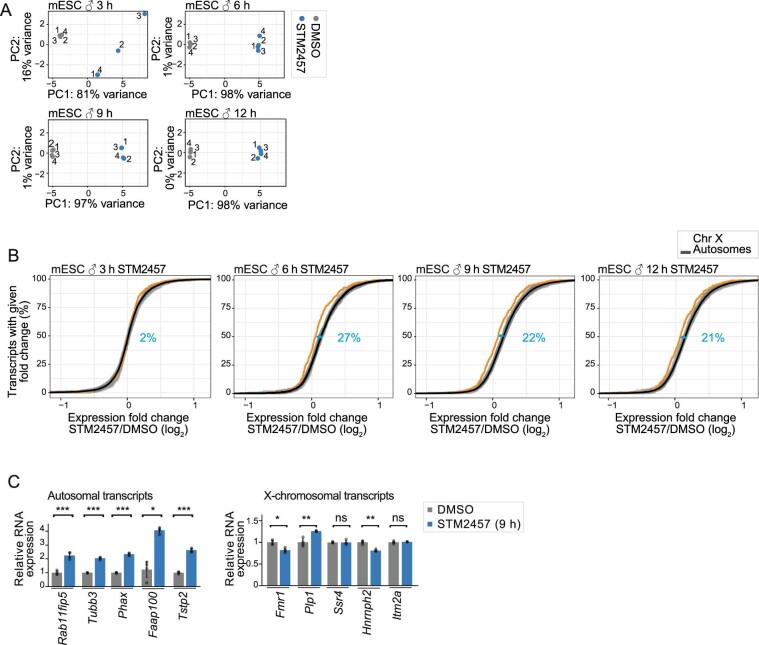
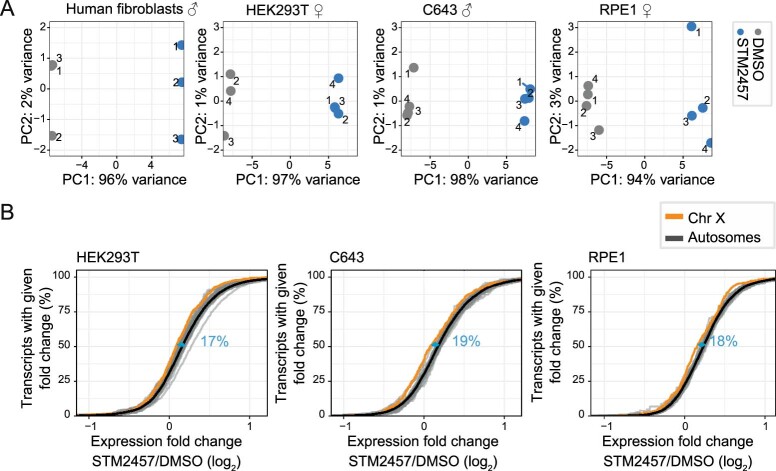
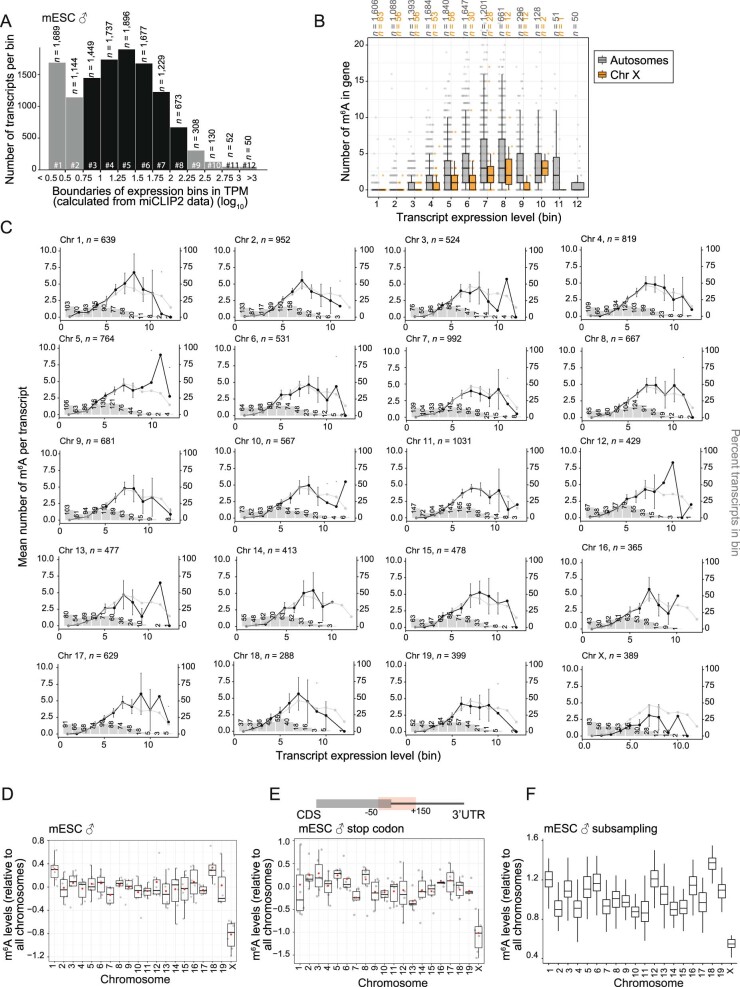
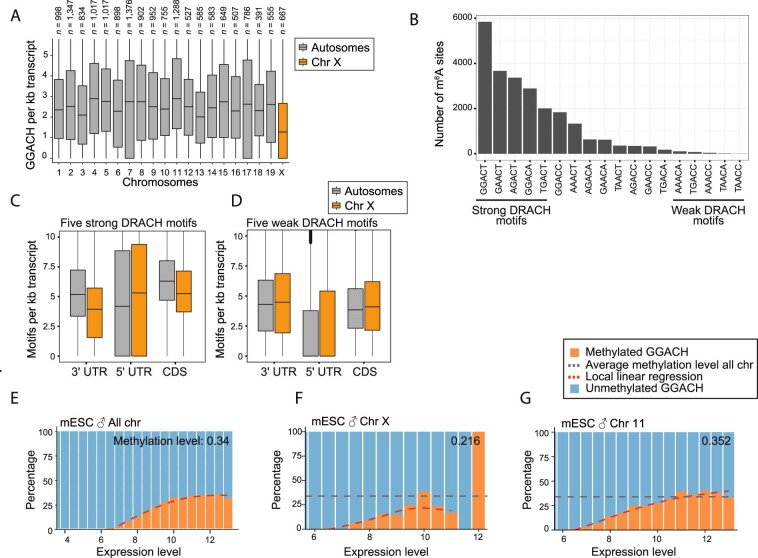
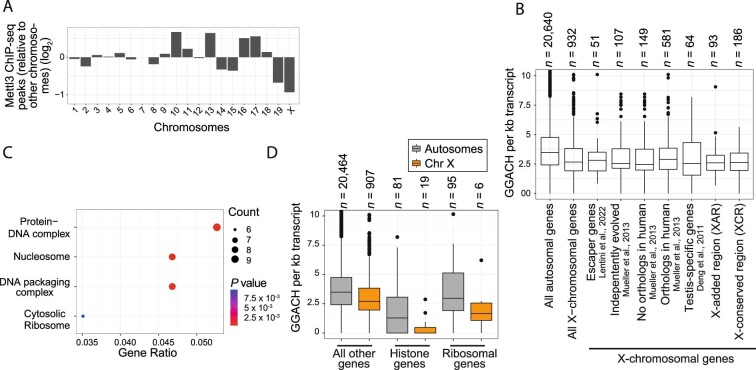
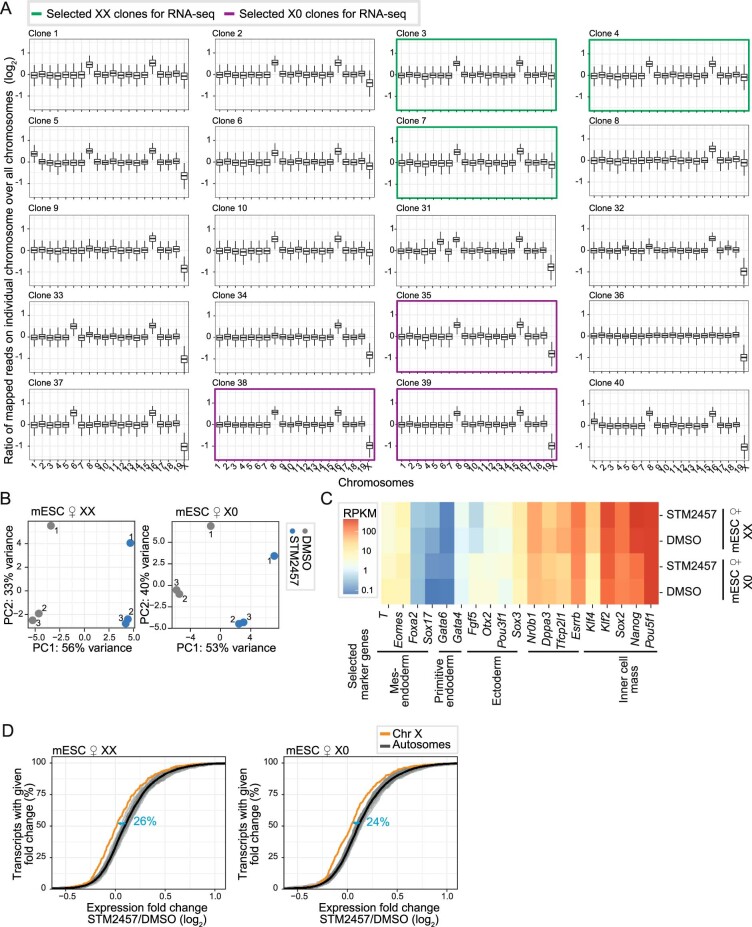
In mammals, X-chromosomal genes are expressed from a single copy since males (XY) possess a single X chromosome, while females (XX) undergo X inactivation. To compensate for this reduction in dosage compared with two active copies of autosomes, it has been proposed that genes from the active X chromosome exhibit dosage compensation. However, the existence and mechanisms of X-to-autosome dosage compensation are still under debate. Here we show that X-chromosomal transcripts have fewer m6A modifications and are more stable than their autosomal counterparts. Acute depletion of m6A selectively stabilizes autosomal transcripts, resulting in perturbed dosage compensation in mouse embryonic stem cells. We propose that higher stability of X-chromosomal transcripts is directed by lower levels of m6A, indicating that mammalian dosage compensation is partly regulated by epitranscriptomic RNA modifications.
© 2023. The Author(s).
Conflict of interest statement
O.R. is an employee of STORM Therapeutics Ltd. The other authors declare that they have no competing interests.
Figures
References
-
- Ohno, S. Sex Chromosomes and Sex-Linked Genes (Springer, 1966).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
