

This is a preprint.
The landscape of tolerated genetic variation in humans and primates
- PMID: 37205491
- PMCID: PMC10187174
- DOI: 10.1101/2023.05.01.538953
The landscape of tolerated genetic variation in humans and primates
Update in
-
The landscape of tolerated genetic variation in humans and primates.Science. 2023 Jun 2;380(6648):eabn8153. doi: 10.1126/science.abn8197. Epub 2023 Jun 2. Science. 2023. PMID: 37262156 Free PMC article.
Abstract
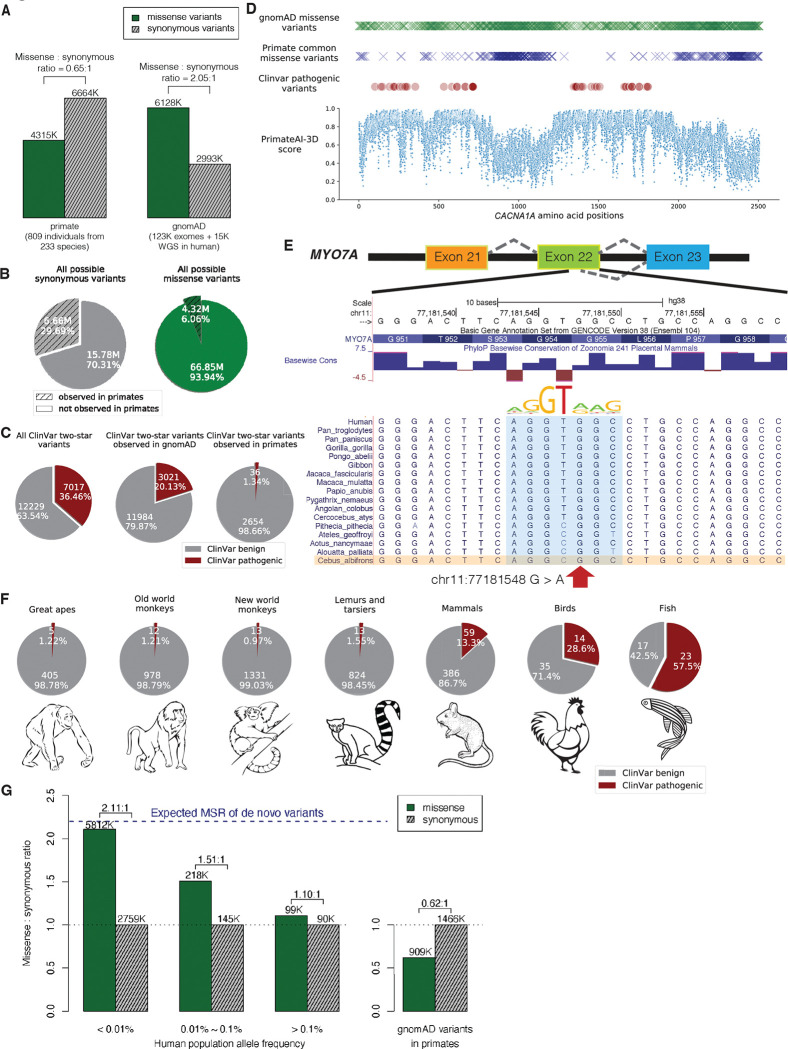
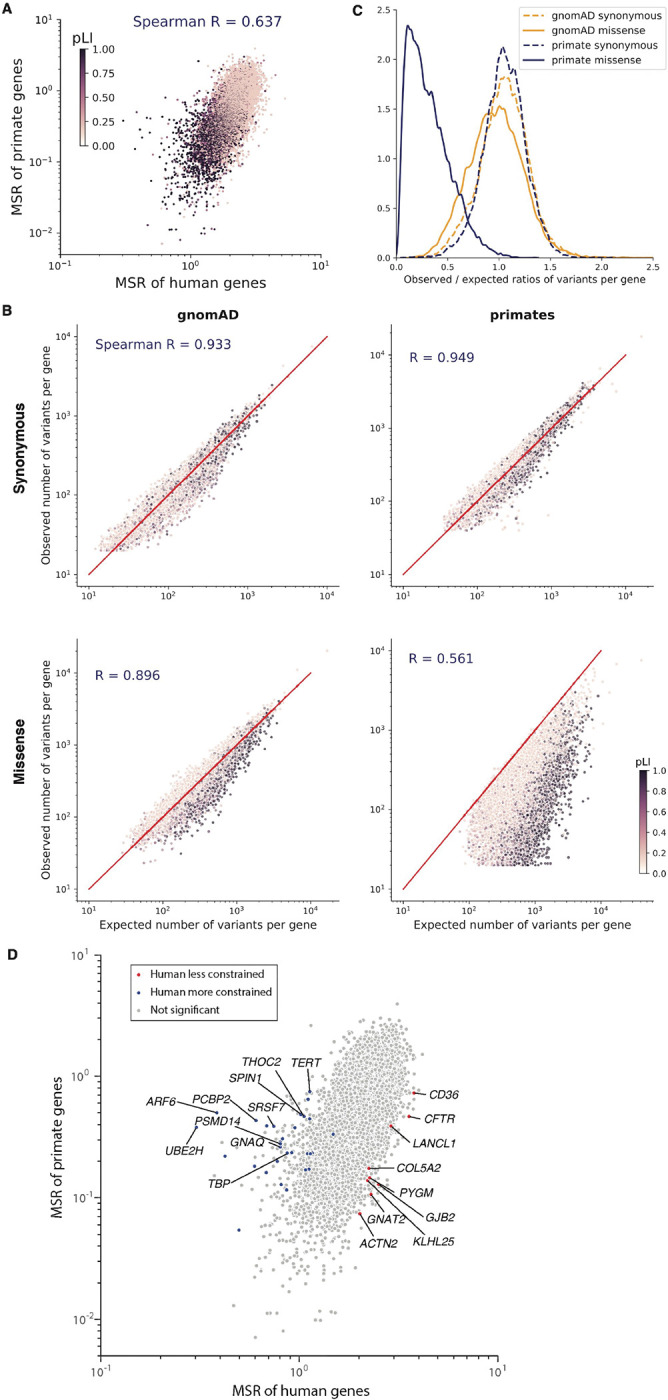
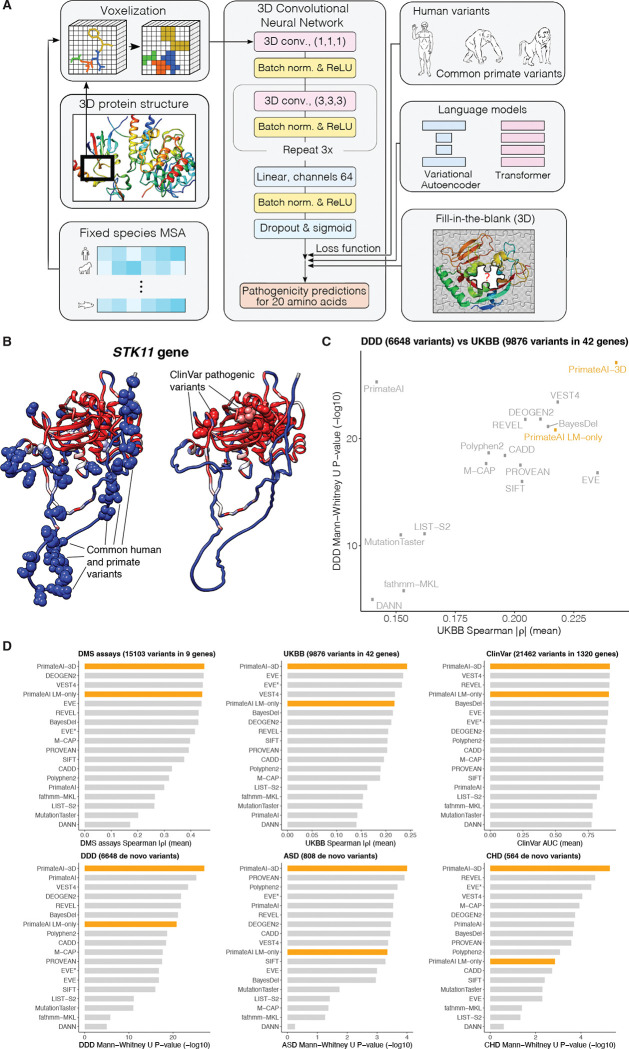
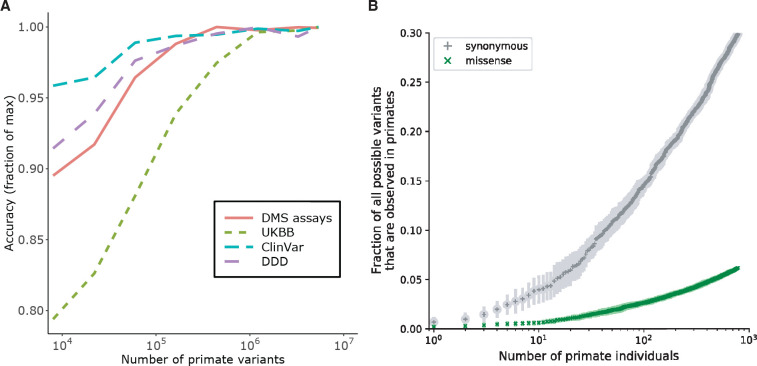
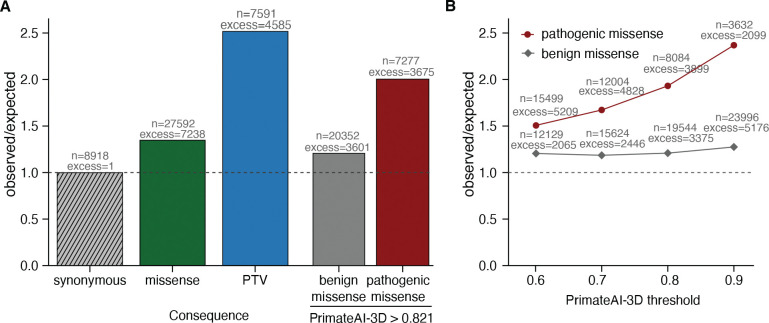
Personalized genome sequencing has revealed millions of genetic differences between individuals, but our understanding of their clinical relevance remains largely incomplete. To systematically decipher the effects of human genetic variants, we obtained whole genome sequencing data for 809 individuals from 233 primate species, and identified 4.3 million common protein-altering variants with orthologs in human. We show that these variants can be inferred to have non-deleterious effects in human based on their presence at high allele frequencies in other primate populations. We use this resource to classify 6% of all possible human protein-altering variants as likely benign and impute the pathogenicity of the remaining 94% of variants with deep learning, achieving state-of-the-art accuracy for diagnosing pathogenic variants in patients with genetic diseases.
One sentence summary: Deep learning classifier trained on 4.3 million common primate missense variants predicts variant pathogenicity in humans.
Conflict of interest statement
Figures
References
-
- Nussbaum R. L., Rehm H. L., ClinGen, ClinGen and Genetic Testing. N Engl J Med 373, 1379 (2015). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources