

The phospholipid transporter PITPNC1 links KRAS to MYC to prevent autophagy in lung and pancreatic cancer
- PMID: 37210549
- PMCID: PMC10199551
- DOI: 10.1186/s12943-023-01788-w
The phospholipid transporter PITPNC1 links KRAS to MYC to prevent autophagy in lung and pancreatic cancer
Erratum in
-
Correction: The phospholipid transporter PITPNC1 links KRAS to MYC to prevent autophagy in lung and pancreatic cancer.Mol Cancer. 2023 Jun 16;22(1):97. doi: 10.1186/s12943-023-01795-x. Mol Cancer. 2023. PMID: 37328838 Free PMC article. No abstract available.
Abstract
Background: The discovery of functionally relevant KRAS effectors in lung and pancreatic ductal adenocarcinoma (LUAD and PDAC) may yield novel molecular targets or mechanisms amenable to inhibition strategies. Phospholipids availability has been appreciated as a mechanism to modulate KRAS oncogenic potential. Thus, phospholipid transporters may play a functional role in KRAS-driven oncogenesis. Here, we identified and systematically studied the phospholipid transporter PITPNC1 and its controlled network in LUAD and PDAC.
Methods: Genetic modulation of KRAS expression as well as pharmacological inhibition of canonical effectors was completed. PITPNC1 genetic depletion was performed in in vitro and in vivo LUAD and PDAC models. PITPNC1-deficient cells were RNA sequenced, and Gene Ontology and enrichment analyses were applied to the output data. Protein-based biochemical and subcellular localization assays were run to investigate PITPNC1-regulated pathways. A drug repurposing approach was used to predict surrogate PITPNC1 inhibitors that were tested in combination with KRASG12C inhibitors in 2D, 3D, and in vivo models.
Results: PITPNC1 was increased in human LUAD and PDAC, and associated with poor patients' survival. PITPNC1 was regulated by KRAS through MEK1/2 and JNK1/2. Functional experiments showed PITPNC1 requirement for cell proliferation, cell cycle progression and tumour growth. Furthermore, PITPNC1 overexpression enhanced lung colonization and liver metastasis. PITPNC1 regulated a transcriptional signature which highly overlapped with that of KRAS, and controlled mTOR localization via enhanced MYC protein stability to prevent autophagy. JAK2 inhibitors were predicted as putative PITPNC1 inhibitors with antiproliferative effect and their combination with KRASG12C inhibitors elicited a substantial anti-tumour effect in LUAD and PDAC.
Conclusions: Our data highlight the functional and clinical relevance of PITPNC1 in LUAD and PDAC. Moreover, PITPNC1 constitutes a new mechanism linking KRAS to MYC, and controls a druggable transcriptional network for combinatorial treatments.
Keywords: KRAS; LUAD; MYC; PDAC; PITPNC1; Therapy; mTOR.
© 2023. The Author(s).
Conflict of interest statement
F.L and S.V. receive research funding from Roche. C.A. and S.V. hold research grants by Revolution Medicines. S.V. is a consultant for LiberaBio. None of the disclosed sources are related to this work.
Figures


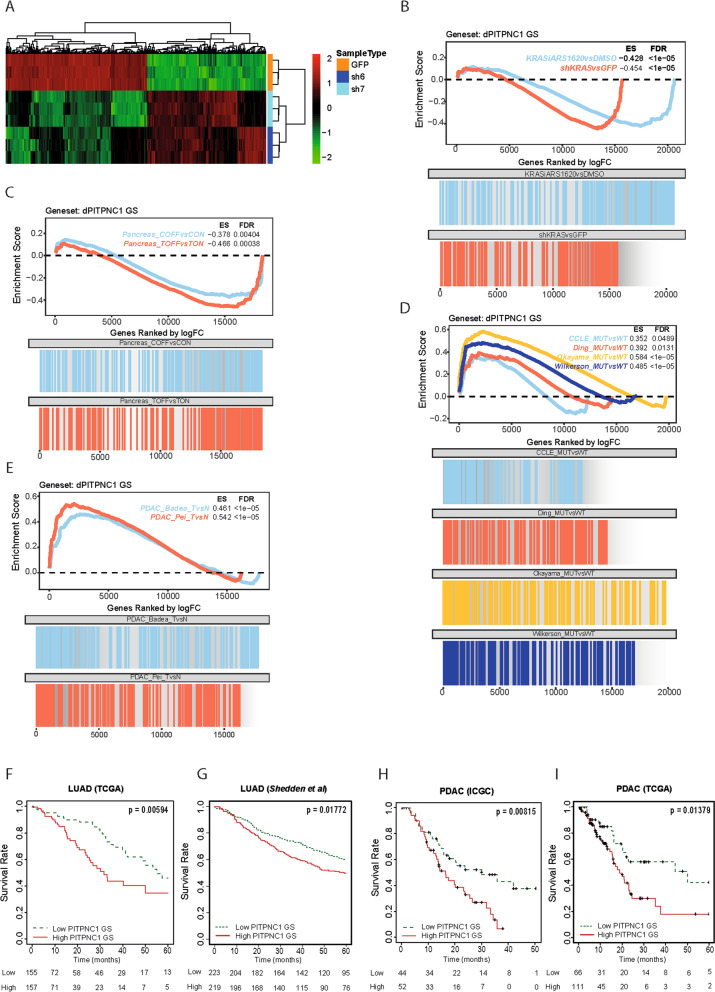




References
-
- Johnson C, Burkhart DL, Haigis KM. Classification of KRAS-activating mutations and the implications for therapeutic intervention. Cancer Discov. 2022;12:913–923. doi: 10.1158/2159-8290.CD-22-0035. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
