

Polyphyllin D punctures hypertrophic lysosomes to reverse drug resistance of hepatocellular carcinoma by targeting acid sphingomyelinase
- PMID: 37211762
- PMCID: PMC10362416
- DOI: 10.1016/j.ymthe.2023.05.015
Polyphyllin D punctures hypertrophic lysosomes to reverse drug resistance of hepatocellular carcinoma by targeting acid sphingomyelinase
Abstract
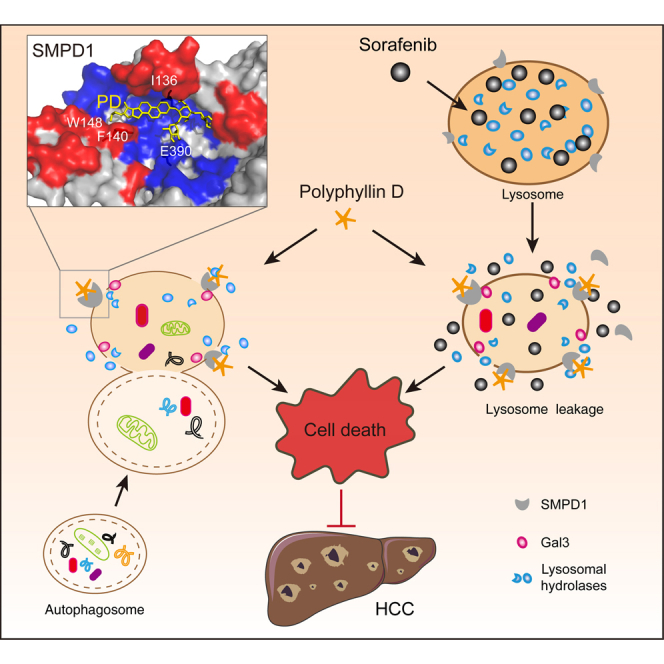
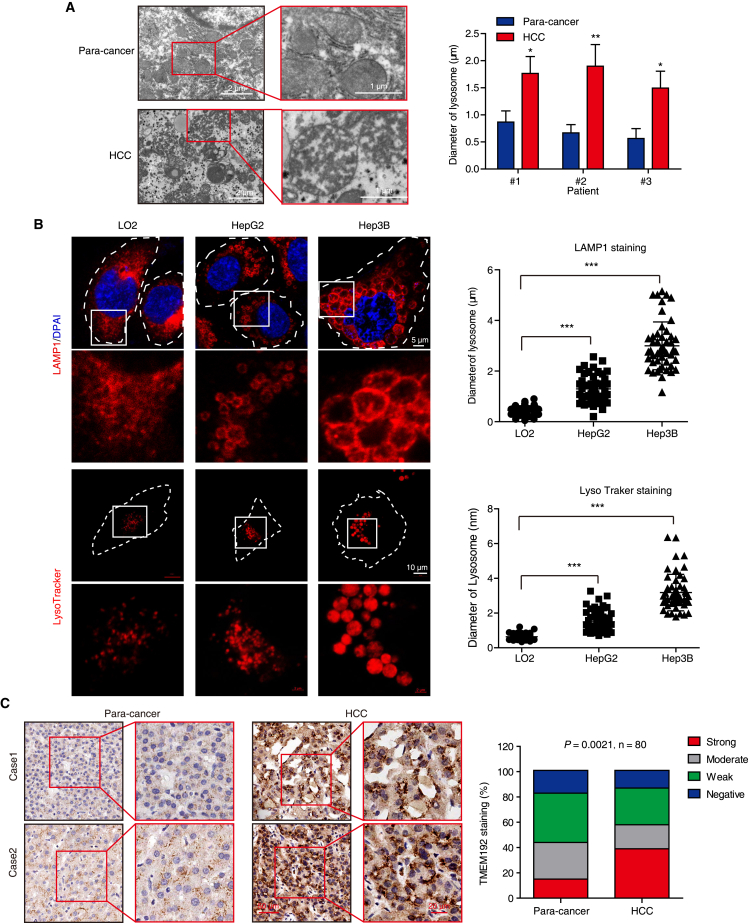
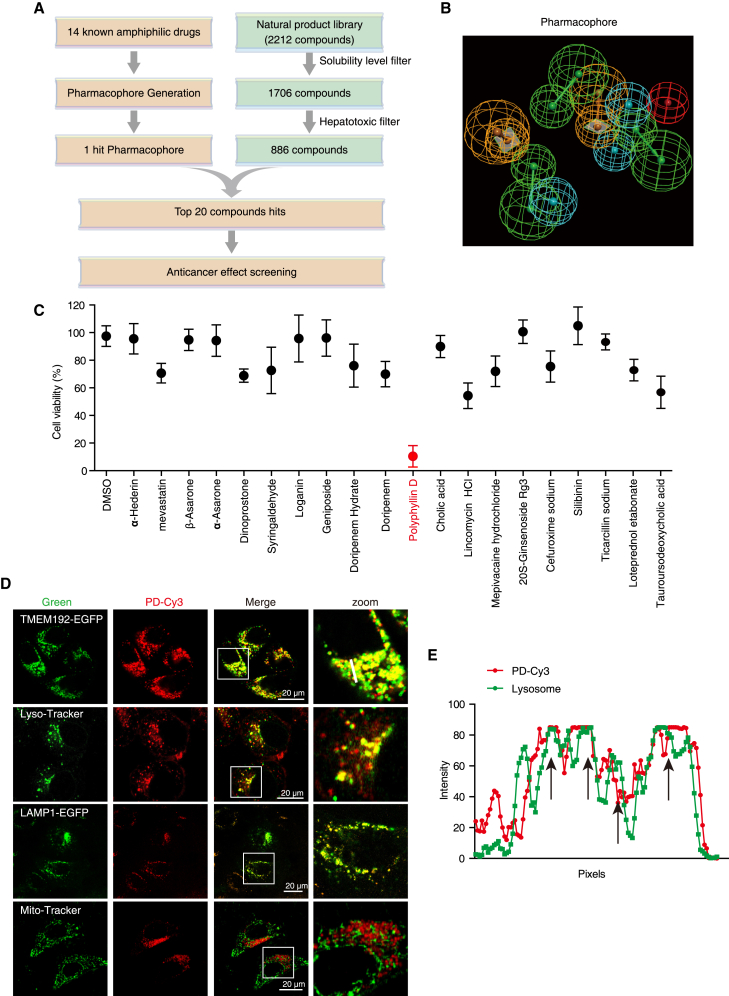
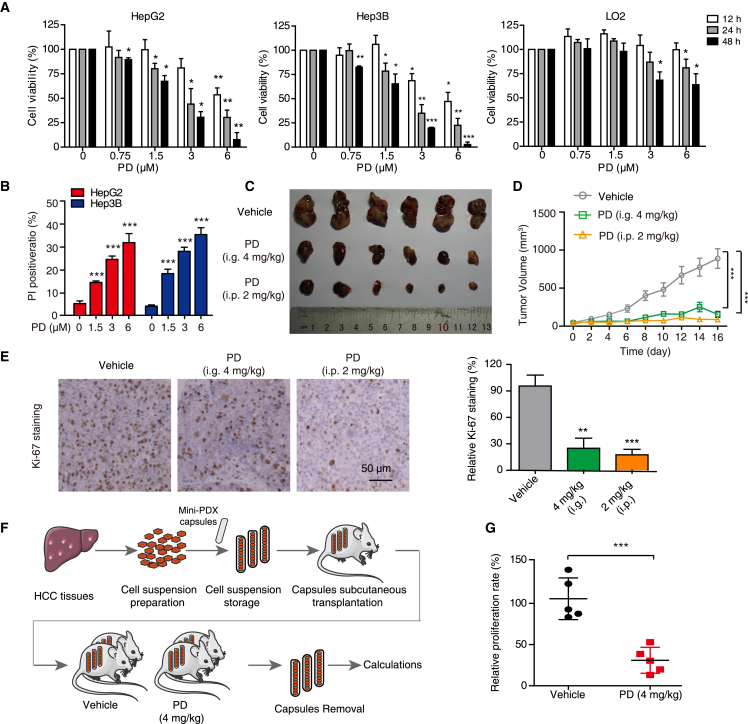
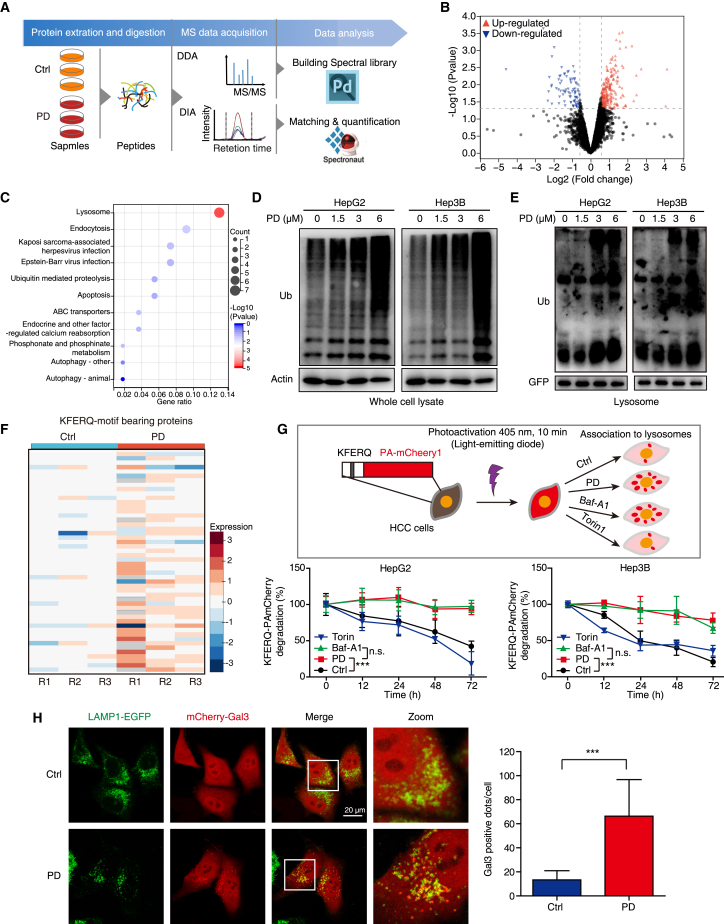
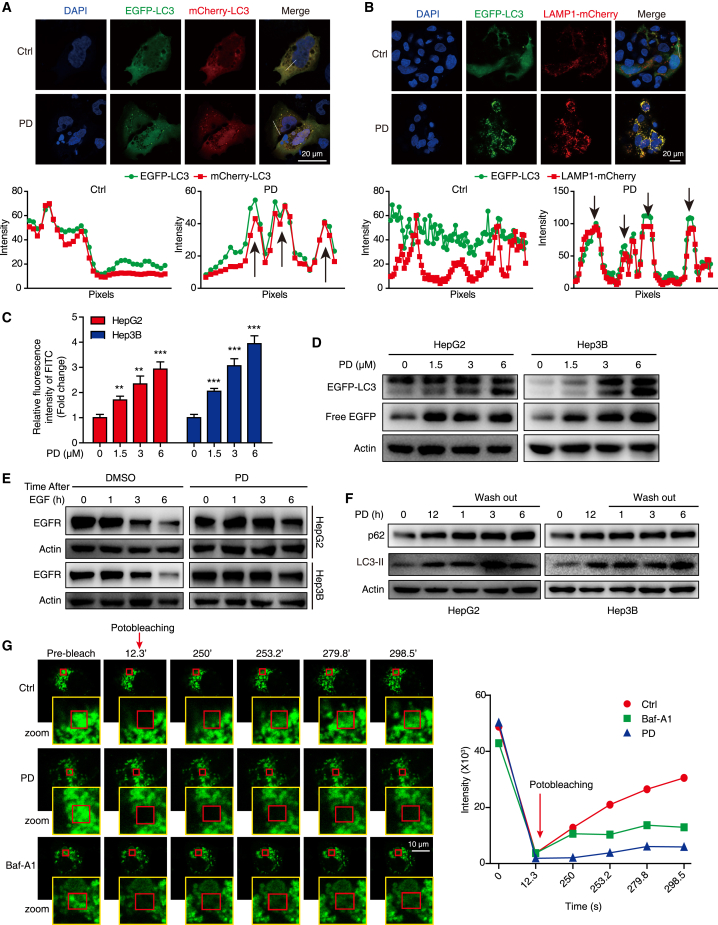
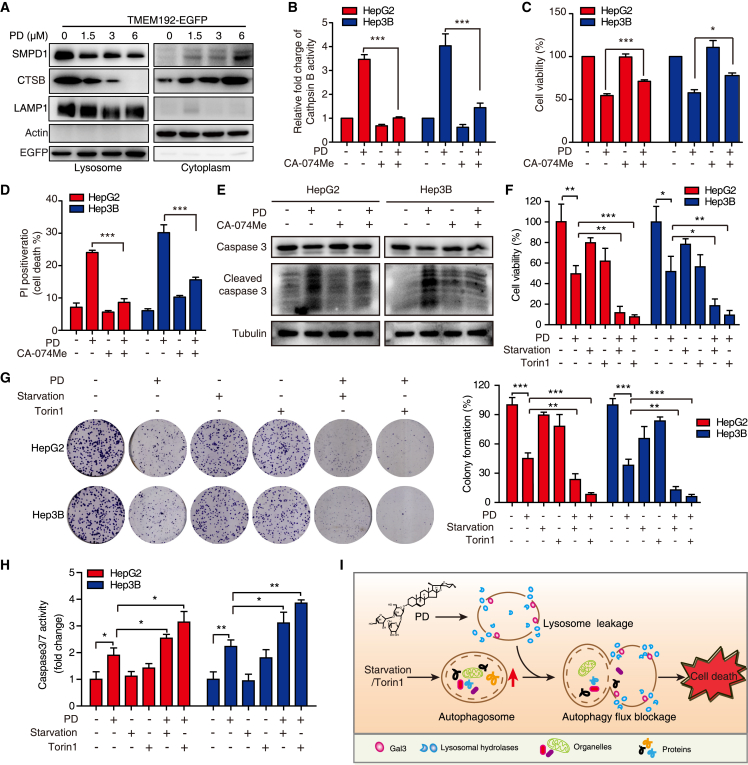
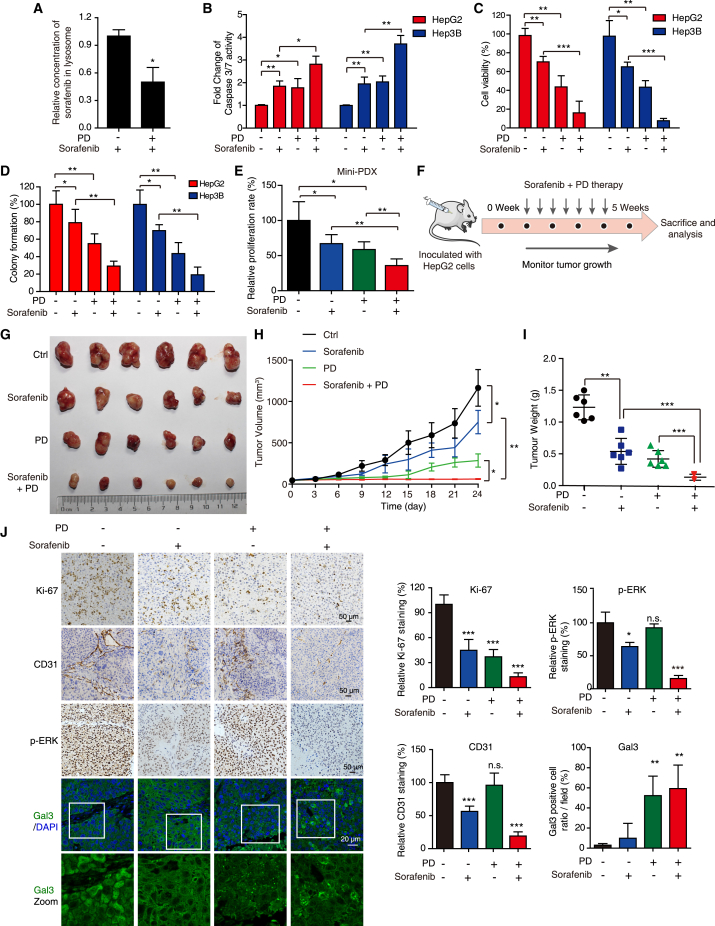
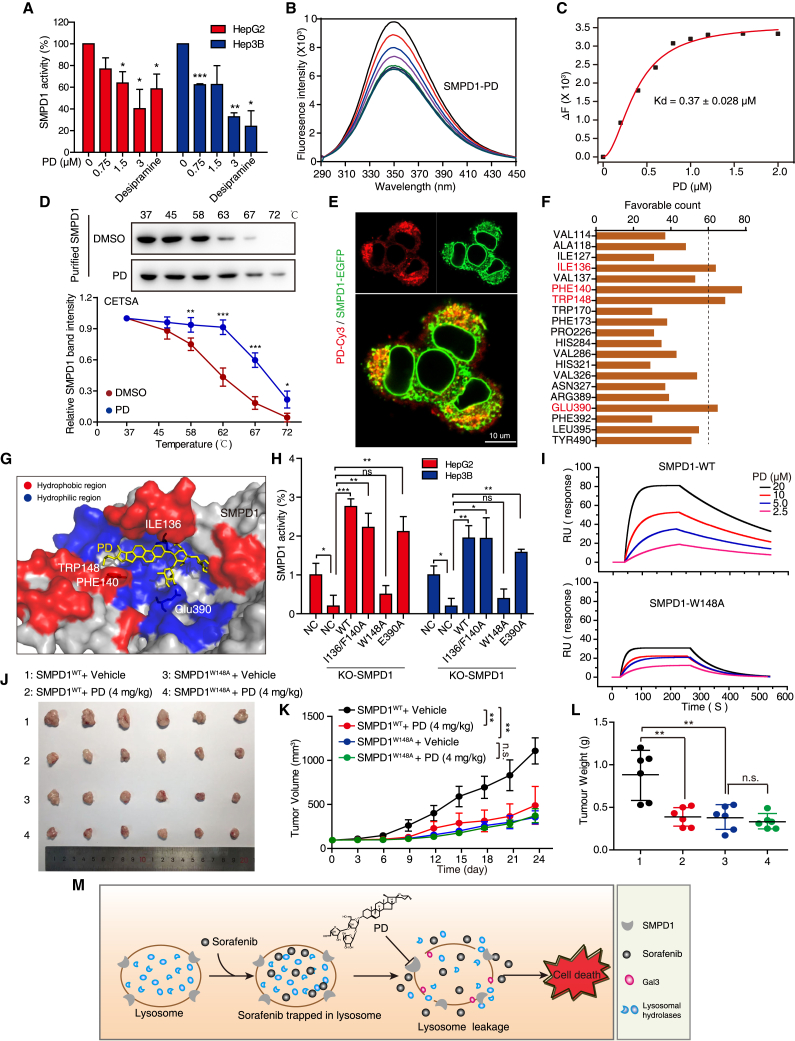
Hypertrophic lysosomes are critical for tumor progression and drug resistance; however, effective and specific lysosome-targeting compounds for cancer therapy are lacking. Here we conducted a lysosomotropic pharmacophore-based in silico screen in a natural product library (2,212 compounds), and identified polyphyllin D (PD) as a novel lysosome-targeted compound. PD treatment was found to cause lysosomal damage, as evidenced by the blockade of autophagic flux, loss of lysophagy, and the release of lysosomal contents, thus exhibiting anticancer effects on hepatocellular carcinoma (HCC) cell both in vitro and in vivo. Closer mechanistic examination revealed that PD suppressed the activity of acid sphingomyelinase (SMPD1), a lysosomal phosphodieserase that catalyzes the hydrolysis of sphingomyelin to produce ceramide and phosphocholine, by directly occupying its surface groove, with Trp148 in SMPD1 acting as a major binding residue; this suppression of SMPD1 activity irreversibly triggers lysosomal injury and initiates lysosome-dependent cell death. Furthermore, PD-enhanced lysosomal membrane permeabilization to release sorafenib, augmenting the anticancer effect of sorafenib both in vivo and in vitro. Overall, our study suggests that PD can potentially be further developed as a novel autophagy inhibitor, and a combination of PD with classical chemotherapeutic anticancer drugs could represent a novel therapeutic strategy for HCC intervention.
Keywords: hepatocellular carcinoma; lysophagy; lysosomal cell death; lysosome hypertrophy; polyphyllin D; sorafenib resistance.
Copyright © 2023 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Serrano-Puebla A., Boya P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 2016;1371:30–44. - PubMed
-
- Gulbins E., Kolesnick R.N. It takes a CAD to kill a tumor cell with a LMP. Cancer Cell. 2013;24:279–281. - PubMed
-
- Mrschtik M., Ryan K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015;282:1858–1870. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
