

Culture-Independent Workflow for Nanopore MinION-Based Sequencing of Influenza A Virus
- PMID: 37212605
- PMCID: PMC10269883
- DOI: 10.1128/spectrum.04946-22
Culture-Independent Workflow for Nanopore MinION-Based Sequencing of Influenza A Virus
Abstract
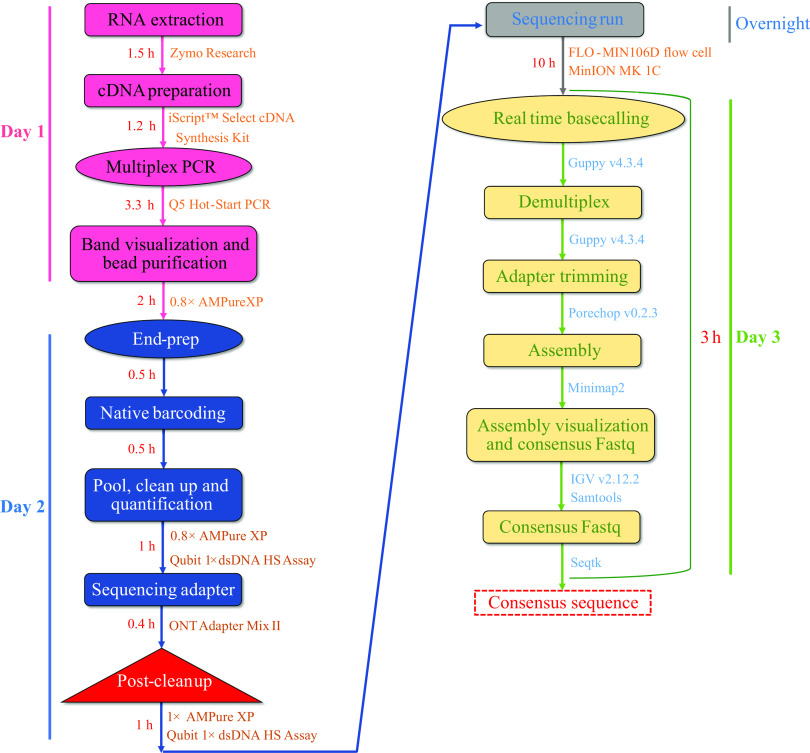
Whole-genome sequencing (WGS) of influenza A virus (IAV) is crucial for identifying diverse subtypes and newly evolved variants and for selecting vaccine strains. In developing countries, where facilities are often inadequate, WGS is challenging to perform using conventional next-generation sequencers. In this study, we established a culture-independent, high-throughput native barcode amplicon sequencing workflow that can sequence all influenza subtypes directly from a clinical specimen. All segments of IAV in 19 clinical specimens, irrespective of their subtypes, were amplified simultaneously using a two-step reverse transcriptase PCR (RT-PCR) system. First, the library was prepared using the ligation sequencing kit, barcoded individually using the native barcodes, and sequenced on the MinION MK 1C platform with real-time base-calling. Then, subsequent data analyses were performed with the appropriate tools. WGS of 19 IAV-positive clinical samples was carried out successfully with 100% coverage and 3,975-fold mean coverage for all segments. This easy-to-install and low-cost capacity-building protocol took only 24 h complete from extracting RNA to obtaining finished sequences. Overall, we developed a high-throughput portable sequencing workflow ideal for resource-limited clinical settings, aiding in real-time surveillance, outbreak investigation, and the detection of emerging viruses and genetic reassortment events. However, further evaluation is required to compare its accuracy with other high-throughput sequencing technologies to validate the widespread application of these findings, including WGS from environmental samples. IMPORTANCE The Nanopore MinION-based influenza sequencing approach we are proposing makes it possible to sequence the influenza A virus, irrespective of its diverse serotypes, directly from clinical and environmental swab samples, so that we are not limited to virus culture. This third-generation, portable, multiplexing, and real-time sequencing strategy is highly convenient for local sequencing, particularly in low- and middle-income countries like Bangladesh. Furthermore, the cost-efficient sequencing method could provide new opportunities to respond to the early phase of an influenza pandemic and enable the timely detection of the emerging subtypes in clinical samples. Here, we meticulously described the entire process that might help the researcher who will follow this methodology in the future. Our findings suggest that this proposed method is ideal for clinical and academic settings and will aid in real-time surveillance and in the detection of potential outbreak agents and newly evolved viruses.
Keywords: MinION; clinical specimen; high throughput; influenza; whole genome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, Cohen C, Gran JM, Schanzer D, Cowling BJ, Wu P, Kyncl J, Ang LW, Park M, Redlberger-Fritz M, Yu H, Espenhain L, Krishnan A, Emukule G, van Asten L, Pereira da Silva S, Aungkulanon S, Buchholz U, Widdowson M-A, Bresee JS, Global Seasonal Influenza-associated Mortality Collaborator Network . 2018. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391:1285–1300. doi:10.1016/S0140-6736(17)33293-2. - DOI - PMC - PubMed
-
- Ito Y. 2018. Clinical diagnosis of influenza, p 23–31. In Influenza virus. Springer, New York, NY.
-
- Houlihan CF, Frampton D, Ferns RB, Raffle J, Grant P, Reidy M, Hail L, Thomson K, Mattes F, Kozlakidis Z, Pillay D, Hayward A, Nastouli E. 2018. Use of whole-genome sequencing in the investigation of a nosocomial influenza virus outbreak. J Infect Dis 218:1485–1489. doi:10.1093/infdis/jiy335. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
