

Variable Region Sequences Influence 16S rRNA Performance
- PMID: 37212673
- PMCID: PMC10269663
- DOI: 10.1128/spectrum.01252-23
Variable Region Sequences Influence 16S rRNA Performance
Abstract
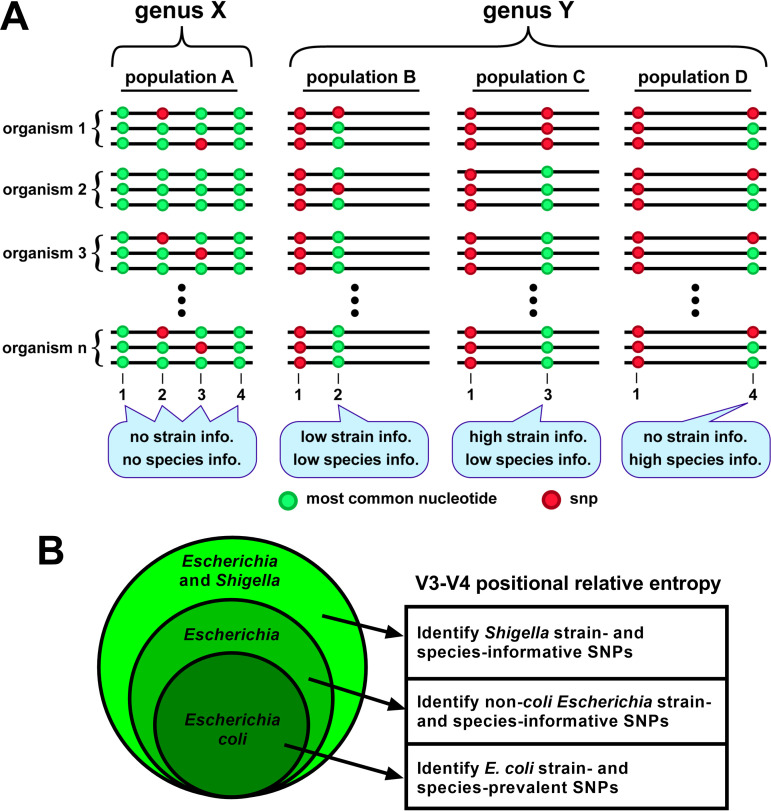
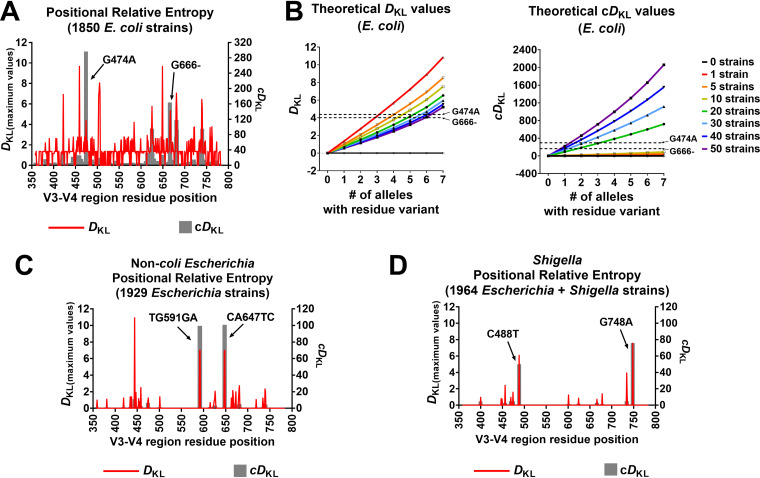
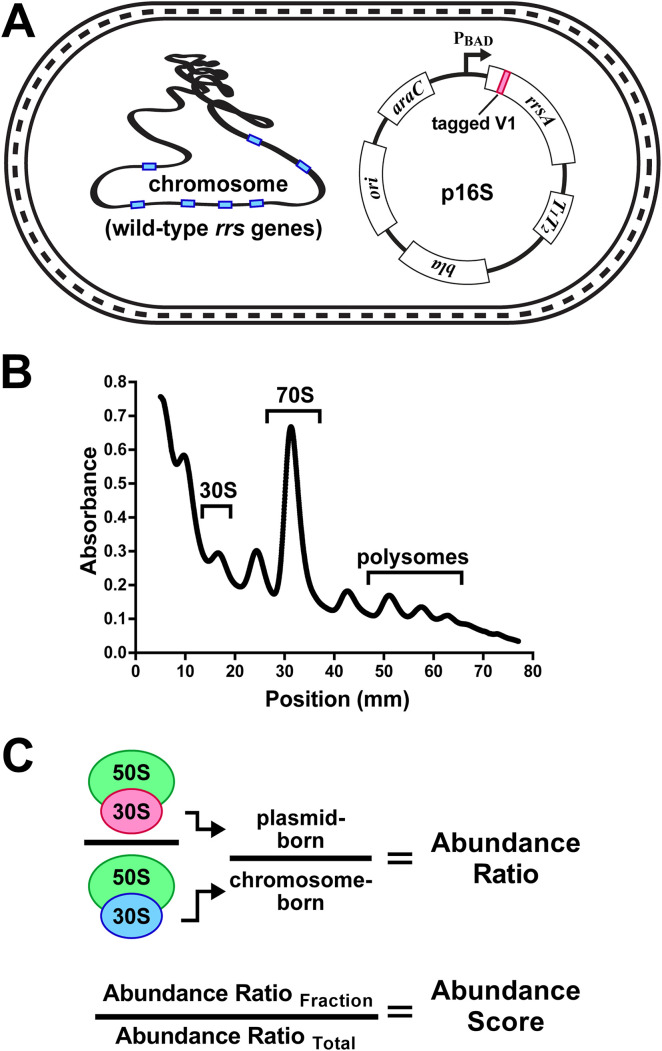
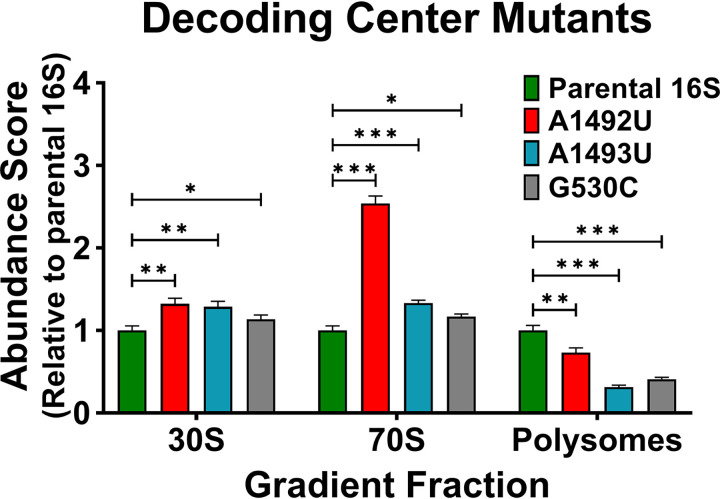
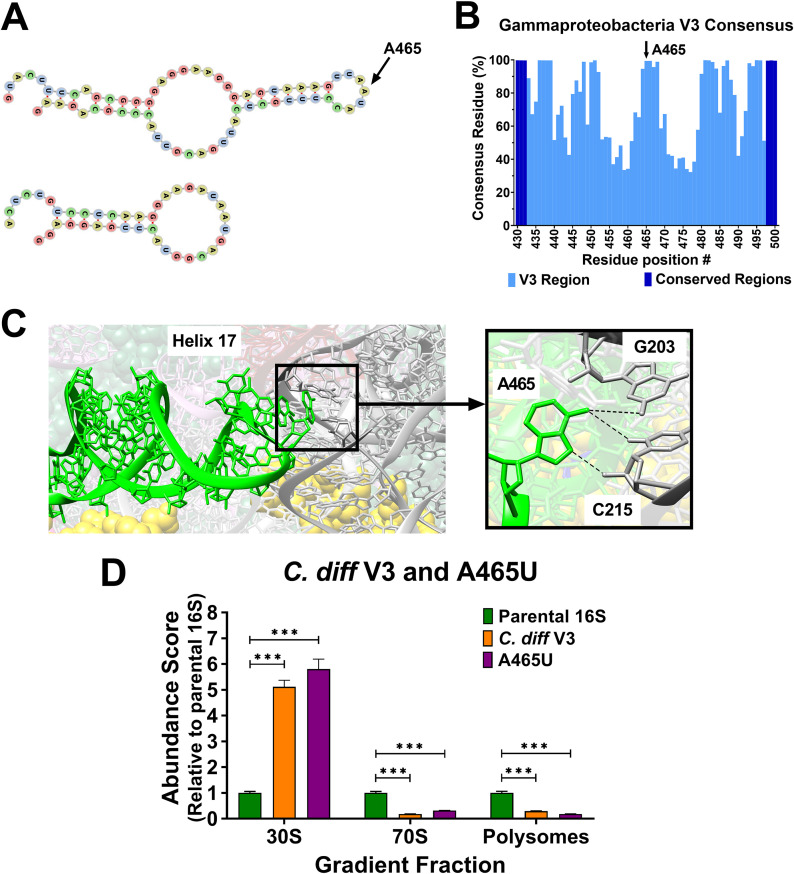
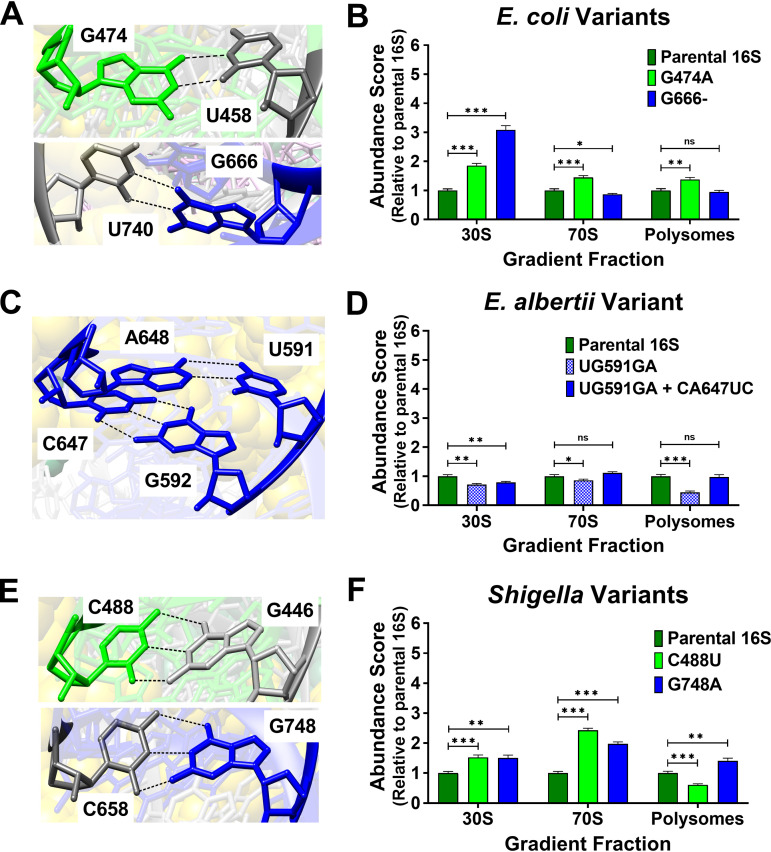
16S rRNA gene sequences are commonly analyzed for taxonomic and phylogenetic studies because they contain variable regions that can help distinguish different genera. However, intra-genus distinction using variable region homology is often impossible due to the high overall sequence identities among closely related species, even though some residues may be conserved within respective species. Using a computational method that included the allelic diversity within individual genomes, we discovered that certain Escherichia and Shigella species can be distinguished by a multi-allelic 16S rRNA variable region single nucleotide polymorphism (SNP). To evaluate the performance of 16S rRNAs with altered variable regions, we developed an in vivo system that measures the acceptance and distribution of variant 16S rRNAs into a large pool of natural versions supporting normal translation and growth. We found that 16S rRNAs containing evolutionarily disparate variable regions were underpopulated both in ribosomes and in active translation pools, even for an SNP. Overall, this study revealed that variable region sequences can substantially influence the performance of 16S rRNAs and that this biological constraint can be leveraged to justify refining taxonomic assignments of variable region sequence data. IMPORTANCE This study reevaluates the notion that 16S rRNA gene variable region sequences are uninformative for intra-genus classification and that single nucleotide variations within them have no consequence to strains that bear them. We demonstrated that the performance of 16S rRNAs in Escherichia coli can be negatively impacted by sequence changes in variable regions, even for single nucleotide changes that are native to closely related Escherichia and Shigella species; thus, biological performance is likely constraining the evolution of variable regions in bacteria. Further, the native nucleotide variations we tested occur in all strains of their respective species and across their multiple 16S rRNA gene copies, suggesting that these species evolved beyond what would be discerned from a consensus sequence comparison. Therefore, this work also reveals that the multiple 16S rRNA gene alleles found in most bacteria can provide more informative phylogenetic and taxonomic detail than a single reference allele.
Keywords: 16S rRNA; relative entropy; ribosome quality; single nucleotide polymorphism; taxonomy; variable region.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
