

Recessive CHRM5 variant as a potential cause of neurogenic bladder
- PMID: 37213061
- PMCID: PMC10527291
- DOI: 10.1002/ajmg.a.63241
Recessive CHRM5 variant as a potential cause of neurogenic bladder
Abstract
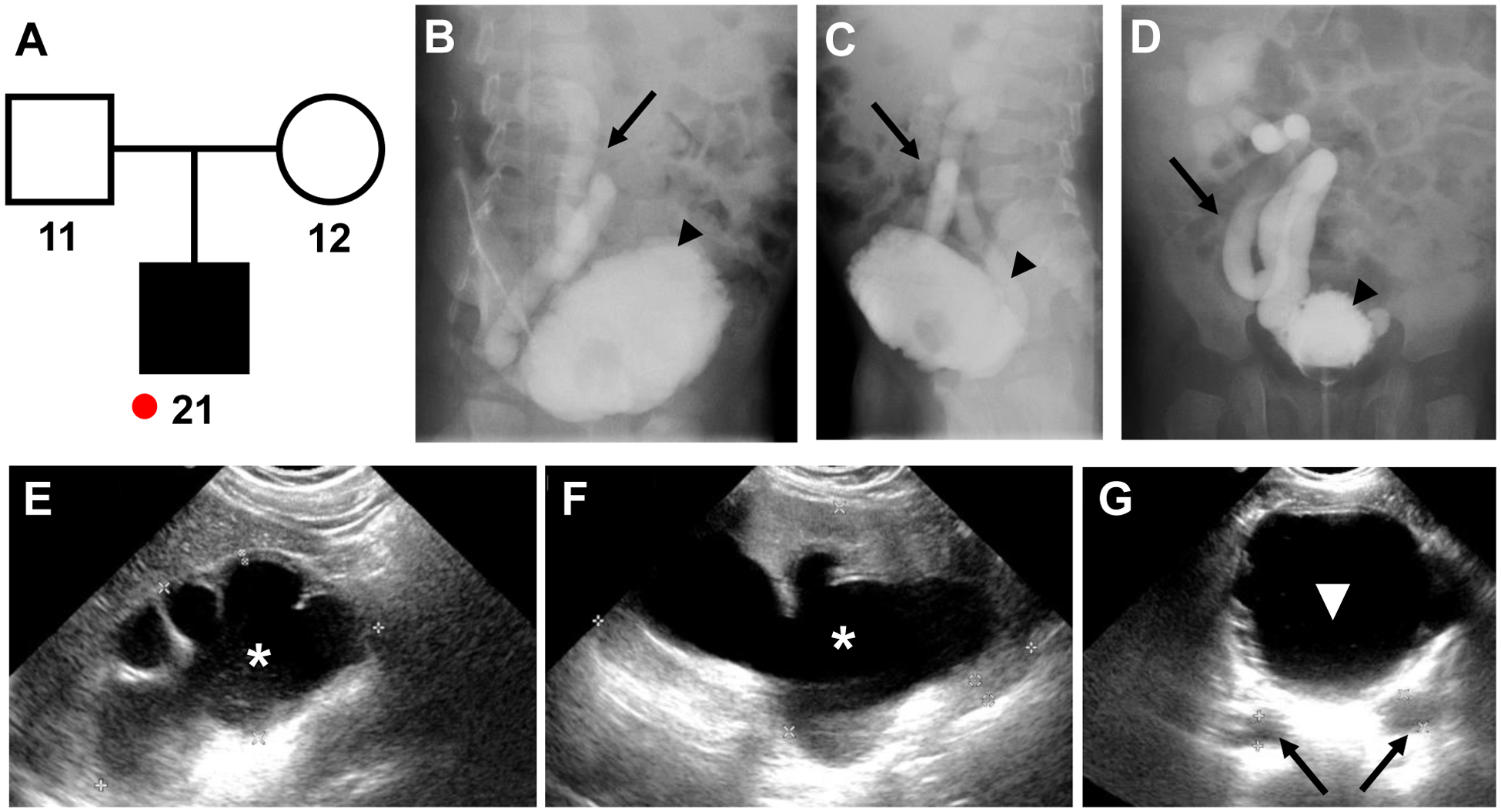
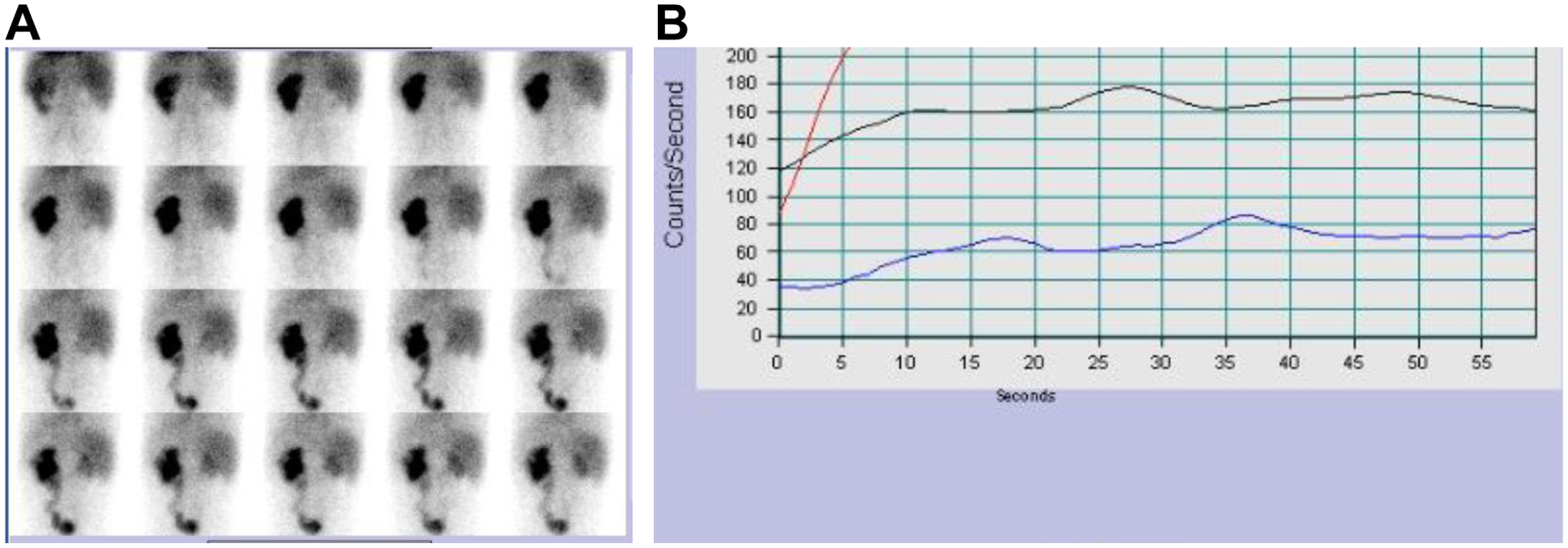
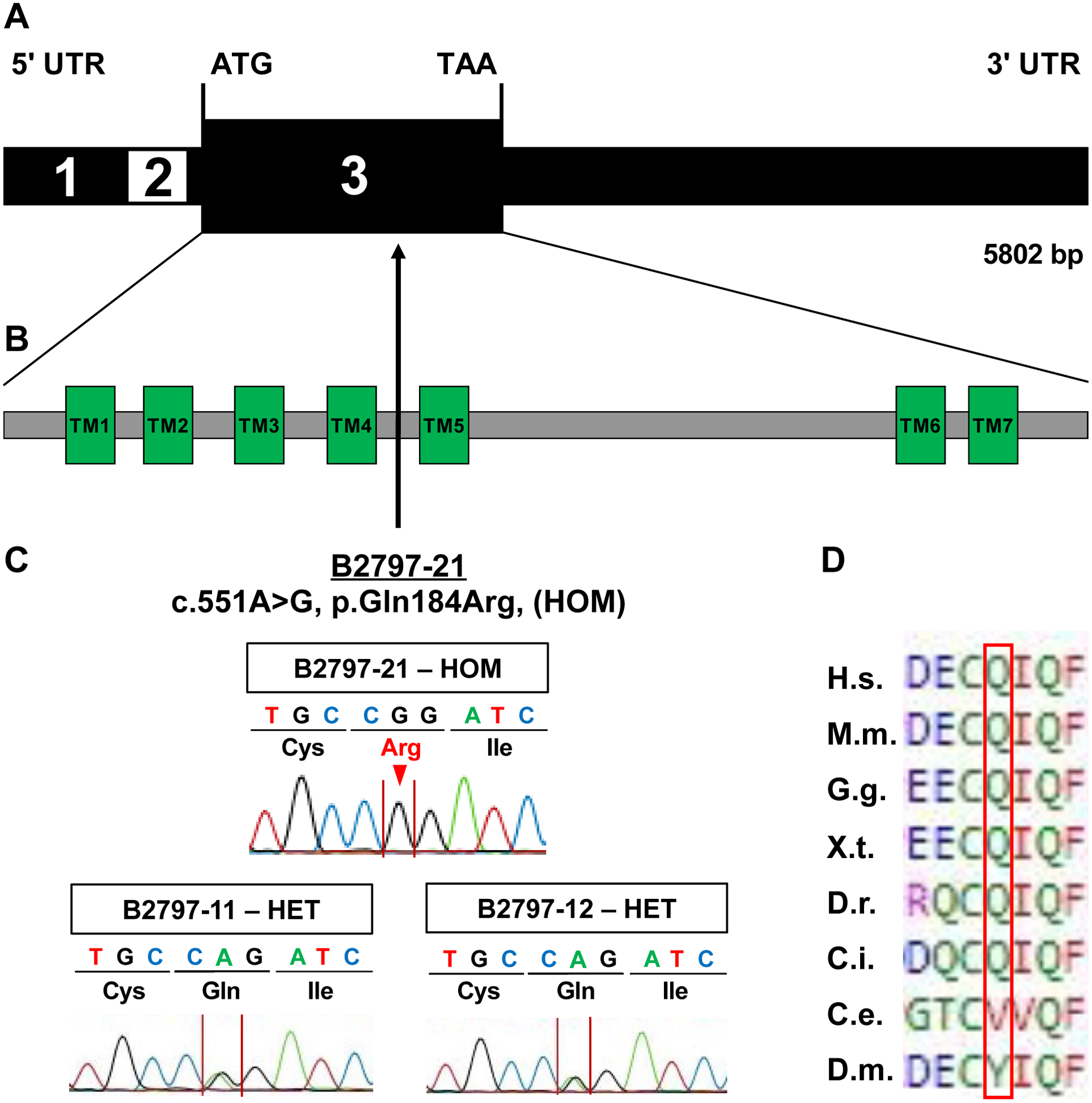
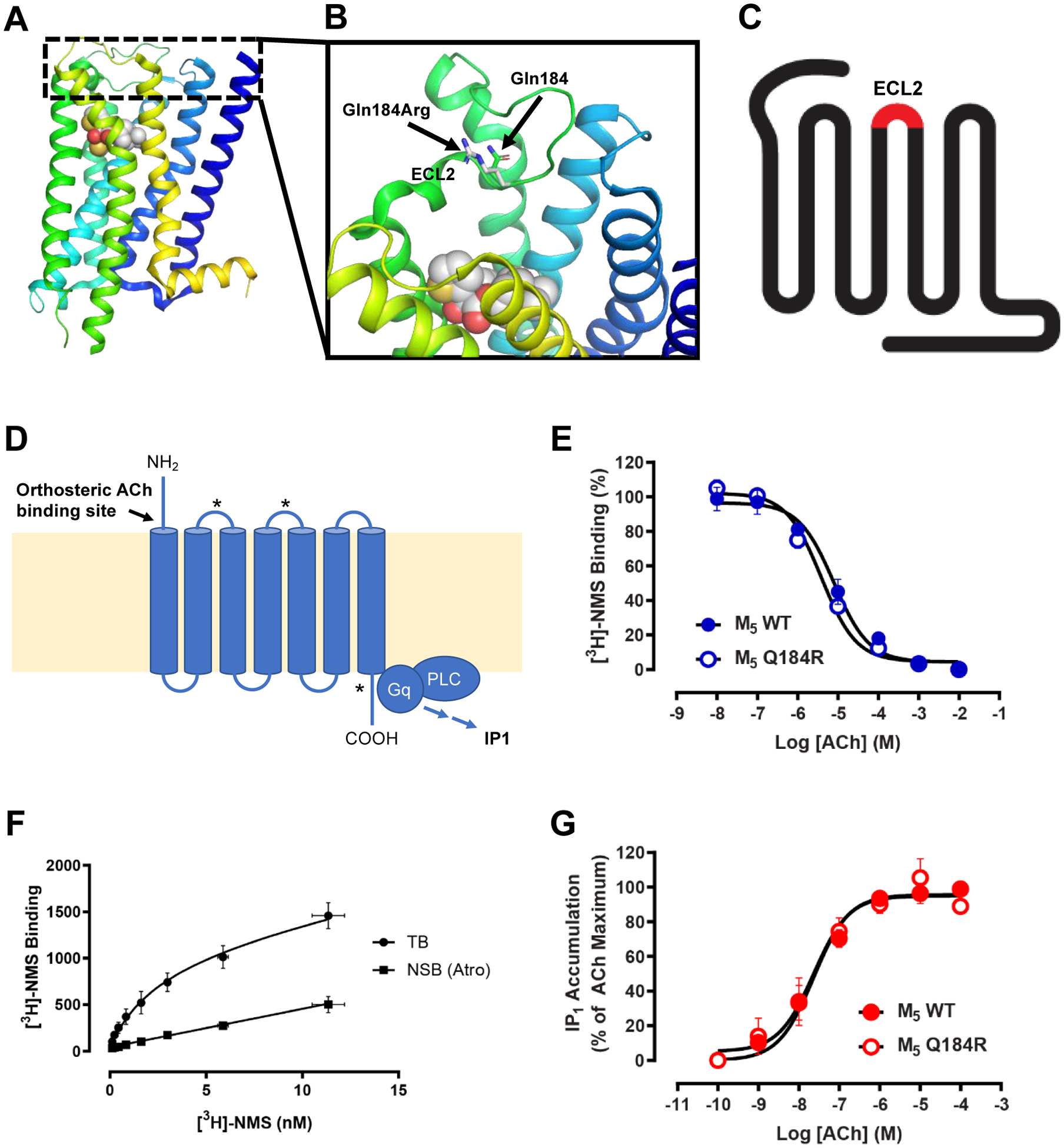
Neurogenic bladder is caused by disruption of neuronal pathways regulating bladder relaxation and contraction. In severe cases, neurogenic bladder can lead to vesicoureteral reflux, hydroureter, and chronic kidney disease. These complications overlap with manifestations of congenital anomalies of the kidney and urinary tract (CAKUT). To identify novel monogenic causes of neurogenic bladder, we applied exome sequencing (ES) to our cohort of families with CAKUT. By ES, we have identified a homozygous missense variant (p.Gln184Arg) in CHRM5 (cholinergic receptor, muscarinic, 5) in a patient with neurogenic bladder and secondary complications of CAKUT. CHRM5 codes for a seven transmembrane-spanning G-protein-coupled muscarinic acetylcholine receptor. CHRM5 is shown to be expressed in murine and human bladder walls and is reported to cause bladder overactivity in Chrm5 knockout mice. We investigated CHRM5 as a potential novel candidate gene for neurogenic bladder with secondary complications of CAKUT. CHRM5 is similar to the cholinergic bladder neuron receptor CHRNA3, which Mann et al. published as the first monogenic cause of neurogenic bladder. However, functional in vitro studies did not reveal evidence to strengthen the status as a candidate gene. Discovering additional families with CHRM5 variants could help to further assess the genes' candidate status.
Keywords: CAKUT; CHRM5; neurogenic bladder; whole-exome sequencing.
© 2023 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest with the research performed.
Figures
Similar articles
-
Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract.J Am Soc Nephrol. 2018 Sep;29(9):2348-2361. doi: 10.1681/ASN.2017121265. Epub 2018 Aug 24. J Am Soc Nephrol. 2018. PMID: 30143558 Free PMC article.
-
Whole-exome sequencing identifies FOXL2, FOXA2 and FOXA3 as candidate genes for monogenic congenital anomalies of the kidneys and urinary tract.Nephrol Dial Transplant. 2022 Sep 22;37(10):1833-1843. doi: 10.1093/ndt/gfab253. Nephrol Dial Transplant. 2022. PMID: 34473308 Free PMC article.
-
Implication of transcription factor FOXD2 dysfunction in syndromic congenital anomalies of the kidney and urinary tract (CAKUT).Kidney Int. 2024 Apr;105(4):844-864. doi: 10.1016/j.kint.2023.11.032. Epub 2023 Dec 26. Kidney Int. 2024. PMID: 38154558 Free PMC article.
-
The genetics and pathogenesis of CAKUT.Nat Rev Nephrol. 2023 Nov;19(11):709-720. doi: 10.1038/s41581-023-00742-9. Epub 2023 Jul 31. Nat Rev Nephrol. 2023. PMID: 37524861 Review.
-
Pathophysiology of Congenital Anomalies of the Kidney and Urinary Tract: A Comprehensive Review.Cells. 2024 Nov 11;13(22):1866. doi: 10.3390/cells13221866. Cells. 2024. PMID: 39594614 Free PMC article. Review.
Cited by
-
Muscarinic control of cardiovascular function in humans: a review of current clinical evidence.Clin Auton Res. 2024 Feb;34(1):31-44. doi: 10.1007/s10286-024-01016-5. Epub 2024 Feb 2. Clin Auton Res. 2024. PMID: 38305989 Free PMC article. Review.
References
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet 12: 745–755. - PubMed
-
- Bschleipfer T, Schukowski K, Weidner W, Grando SA, Schwantes U, Kummer W, Lips KS. 2007. Expression and distribution of cholinergic receptors in the human urothelium. Life Sci 80: 2303–2307. - PubMed
-
- Burger WAC, Gentry PR, Berizzi AE, Vuckovic Z, van der Westhuizen ET, Thompson G, Yeasmin M, Lindsley CW, Sexton PM, Langmead CJ, Tobin AB, Christopoulos A, Valant C, Thal DM. 2021. Identification of a Novel Allosteric Site at the M5 Muscarinic Acetylcholine Receptor. ACS Chem. Neurosci 12: 3112–3123. - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
