

This is a preprint.
A revamped rat reference genome improves the discovery of genetic diversity in laboratory rats
- PMID: 37214860
- PMCID: PMC10197727
- DOI: 10.1101/2023.04.13.536694
A revamped rat reference genome improves the discovery of genetic diversity in laboratory rats
Update in
-
A revamped rat reference genome improves the discovery of genetic diversity in laboratory rats.Cell Genom. 2024 Apr 10;4(4):100527. doi: 10.1016/j.xgen.2024.100527. Epub 2024 Mar 26. Cell Genom. 2024. PMID: 38537634 Free PMC article.
Abstract
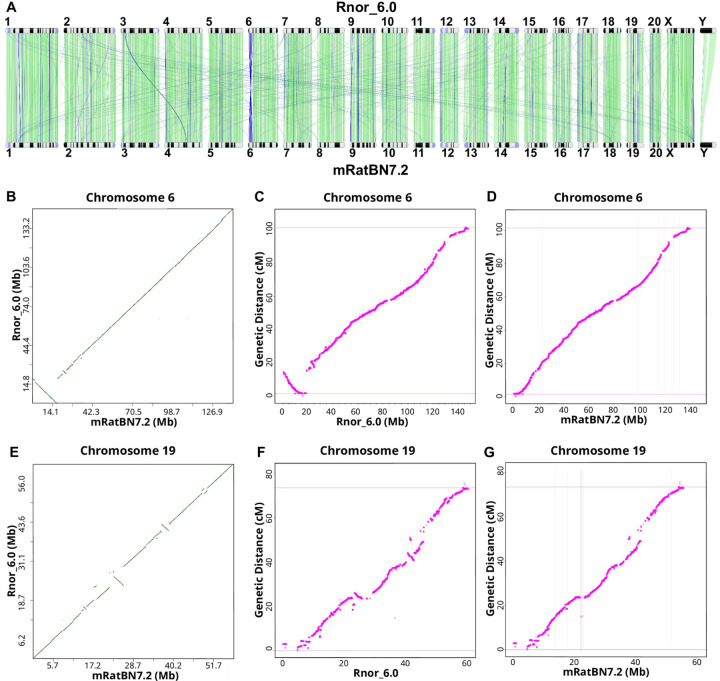
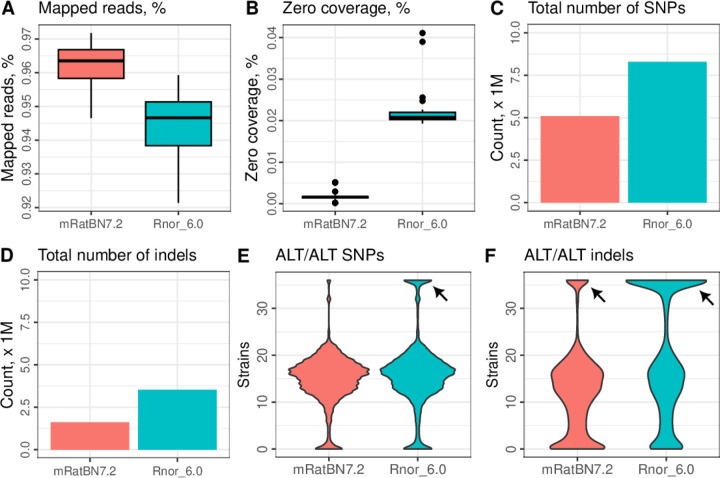
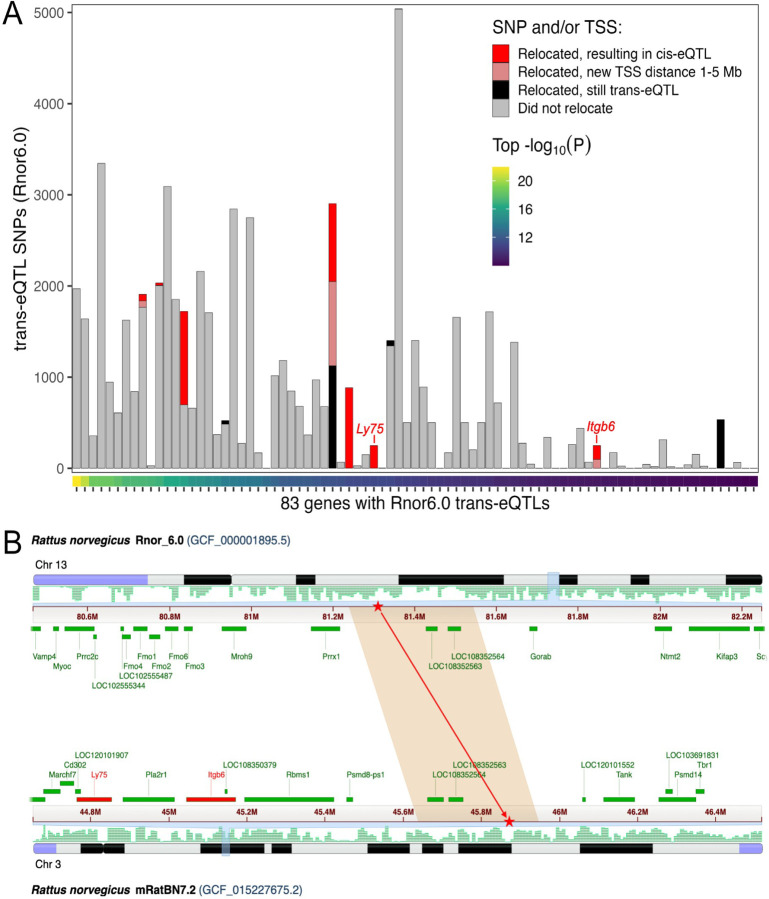
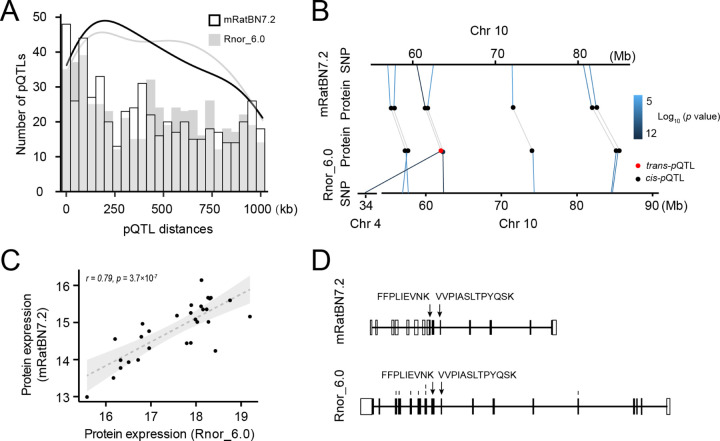
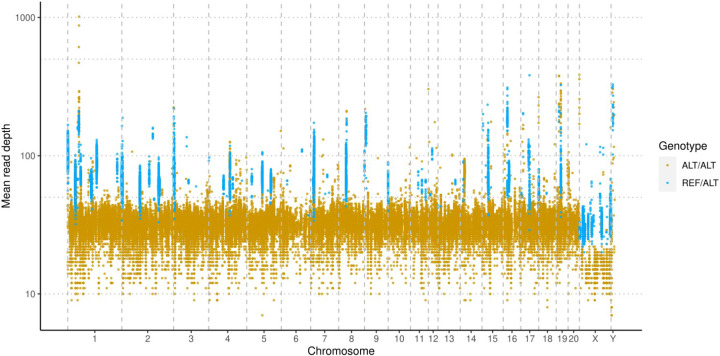
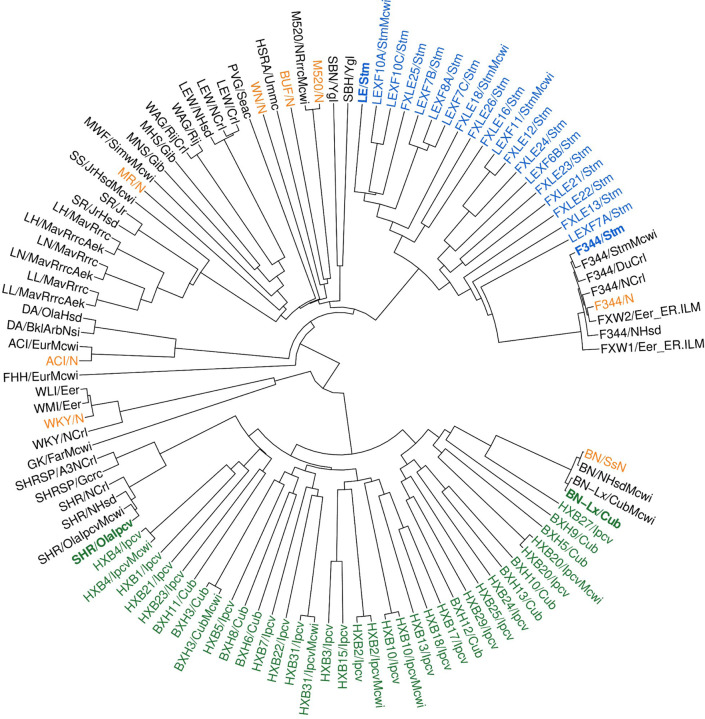
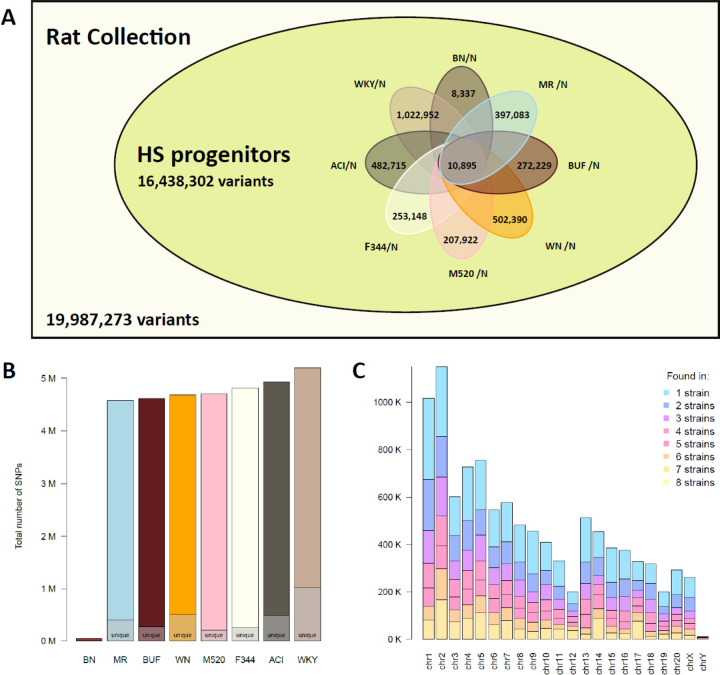
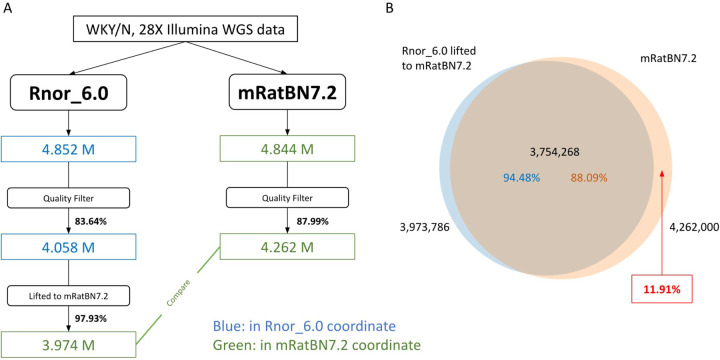
The seventh iteration of the reference genome assembly for Rattus norvegicus-mRatBN7.2-corrects numerous misplaced segments and reduces base-level errors by approximately 9-fold and increases contiguity by 290-fold compared to its predecessor. Gene annotations are now more complete, significantly improving the mapping precision of genomic, transcriptomic, and proteomics data sets. We jointly analyzed 163 short-read whole genome sequencing datasets representing 120 laboratory rat strains and substrains using mRatBN7.2. We defined ~20.0 million sequence variations, of which 18.7 thousand are predicted to potentially impact the function of 6,677 genes. We also generated a new rat genetic map from 1,893 heterogeneous stock rats and annotated transcription start sites and alternative polyadenylation sites. The mRatBN7.2 assembly, along with the extensive analysis of genomic variations among rat strains, enhances our understanding of the rat genome, providing researchers with an expanded resource for studies involving rats.
Keywords: Genetic Map; Heterogeneous Stock; Hybrid Rat Diversity Panel; Inbred Strains; Phylogenetic Tree; Rat; Recombinant Inbred; Reference Genome; Rnor_6.0; mRatBN7.2.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Richter C.P. (1954). The effects of domestication and selection on the behavior of the Norway rat. J. Natl. Cancer Inst. 15, 727–738. - PubMed
-
- Smith J.R., Hayman G.T., Wang S.-J., Laulederkind S.J.F., Hoffman M.J., Kaldunski M.L., Tutaj M., Thota J., Nalabolu H.S., Ellanki S.L.R., et al. (2020). The Year of the Rat: The Rat Genome Database at 20: a multi-species knowledgebase and analysis platform. Nucleic Acids Res. 48, D731–D742. - PMC - PubMed
Publication types
Grants and funding
- R01 GM140287/GM/NIGMS NIH HHS/United States
- P01 HL149620/HL/NHLBI NIH HHS/United States
- U01 DA043098/DA/NIDA NIH HHS/United States
- R01 HL064541/HL/NHLBI NIH HHS/United States
- R24 AA013162/AA/NIAAA NIH HHS/United States
- U24 HG010859/HG/NHGRI NIH HHS/United States
- R21 DK089417/DK/NIDDK NIH HHS/United States
- R01 HL089895/HL/NHLBI NIH HHS/United States
- R01 HG011252/HG/NHGRI NIH HHS/United States
- P30 DA044223/DA/NIDA NIH HHS/United States
- R24 OD024617/OD/NIH HHS/United States
- P50 DA037844/DA/NIDA NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- U01 DA051972/DA/NIDA NIH HHS/United States
- U01 DA050239/DA/NIDA NIH HHS/United States
- R01 DA048017/DA/NIDA NIH HHS/United States
- U01 DA047638/DA/NIDA NIH HHS/United States
- U01 DA051234/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources