

This is a preprint.
Insights from a genome-wide truth set of tandem repeat variation
- PMID: 37214979
- PMCID: PMC10197592
- DOI: 10.1101/2023.05.05.539588
Insights from a genome-wide truth set of tandem repeat variation
Abstract
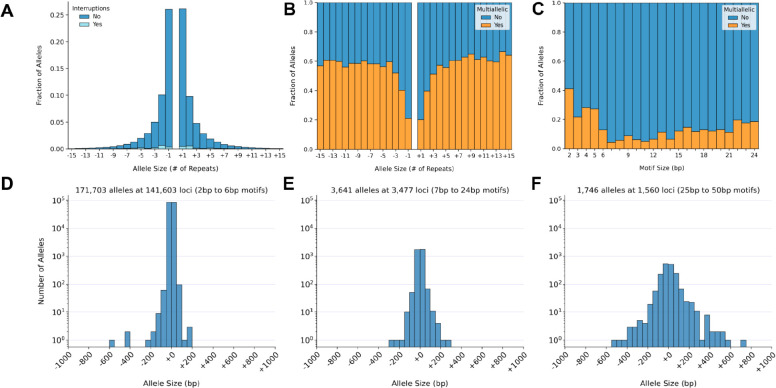
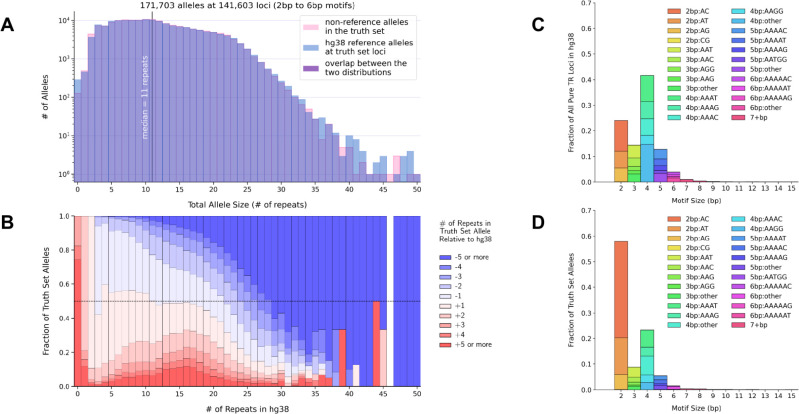
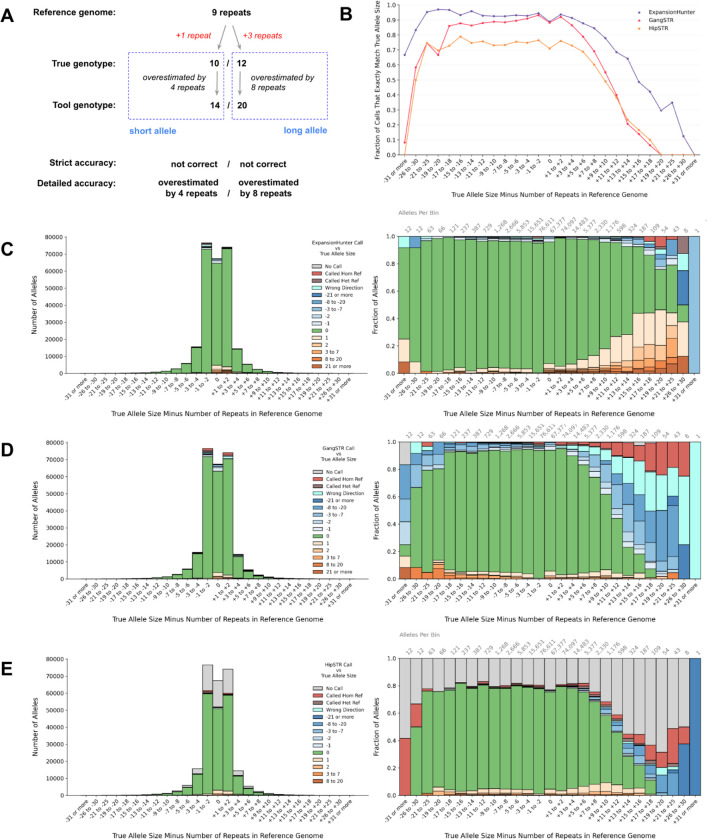
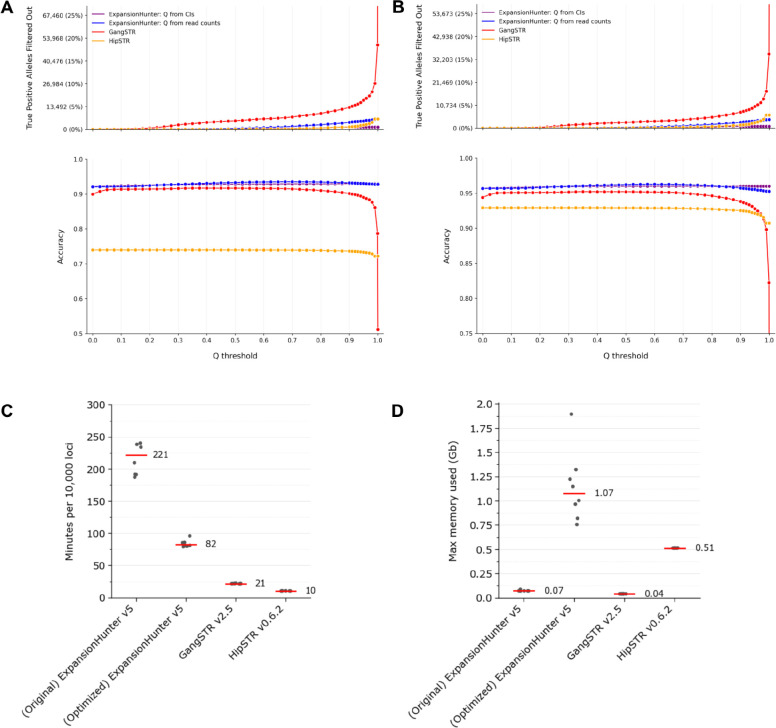
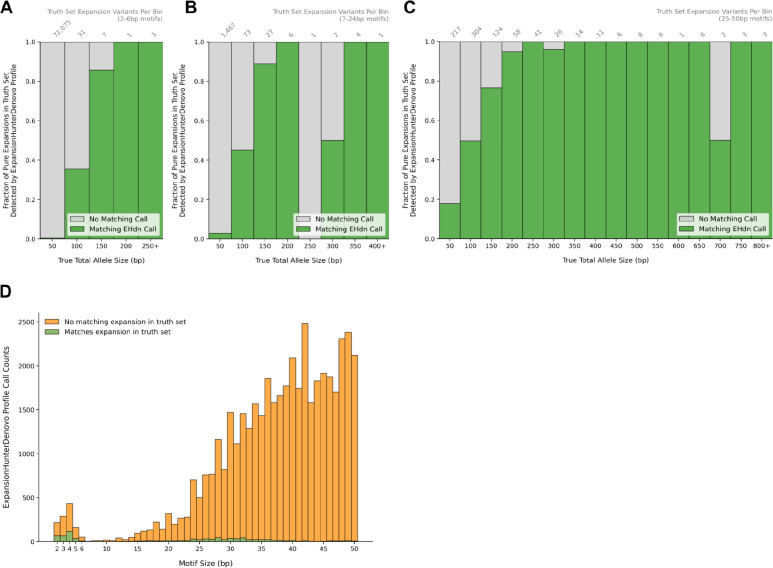
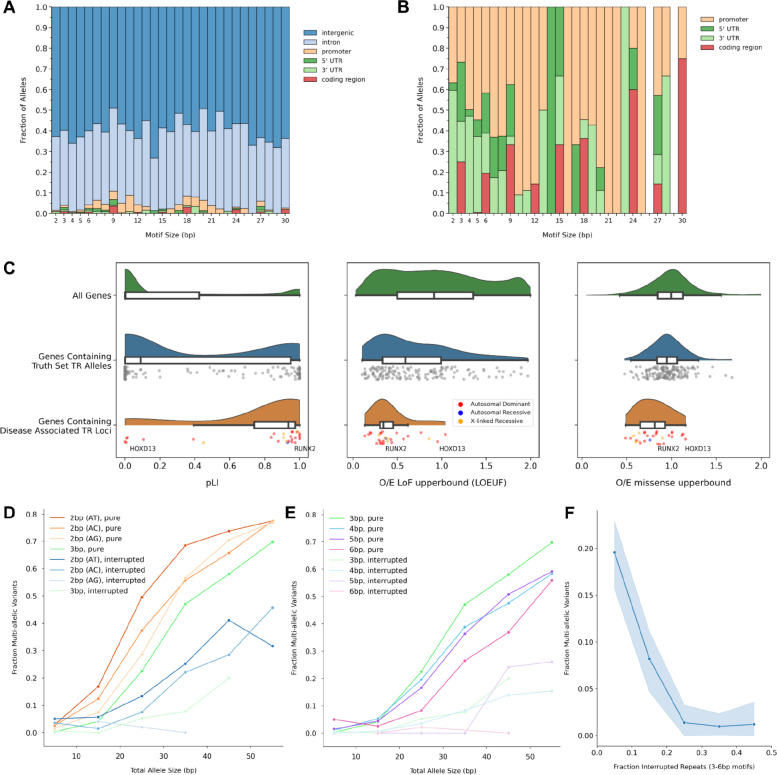
Tools for genotyping tandem repeats (TRs) from short read sequencing data have improved significantly over the past decade. Extensive comparisons of these tools to gold standard diagnostic methods like RP-PCR have confirmed their accuracy for tens to hundreds of well-studied loci. However, a scarcity of high-quality orthogonal truth data limited our ability to measure tool accuracy for the millions of other loci throughout the genome. To address this, we developed a TR truth set based on the Synthetic Diploid Benchmark (SynDip). By identifying the subset of insertions and deletions that represent TR expansions or contractions with motifs between 2 and 50 base pairs, we obtained accurate genotypes for 139,795 pure and 6,845 interrupted repeats in a single diploid sample. Our approach did not require running existing genotyping tools on short read or long read sequencing data and provided an alternative, more accurate view of tandem repeat variation. We applied this truth set to compare the strengths and weaknesses of widely-used tools for genotyping TRs, evaluated the completeness of existing genome-wide TR catalogs, and explored the properties of tandem repeat variation throughout the genome. We found that, without filtering, ExpansionHunter had higher accuracy than GangSTR and HipSTR over a wide range of motifs and allele sizes. Also, when errors in allele size occurred, ExpansionHunter tended to overestimate expansion sizes, while GangSTR tended to underestimate them. Additionally, we saw that widely-used TR catalogs miss between 16% and 41% of variant loci in the truth set. These results suggest that genome-wide analyses would benefit from genotyping a larger set of loci as well as further tool development that builds on the strengths of current algorithms. To that end, we developed a new catalog of 2.8 million loci that captures 95% of variant loci in the truth set, and created a modified version of ExpansionHunter that runs 2 to 3x faster than the original while producing the same output.
Figures

Similar articles
-
Analysis of Tandem Repeats in Short-Read Sequencing Data: From Genotyping Known Pathogenic Repeats to Discovering Novel Expansions.Curr Protoc. 2024 Nov;4(11):e70010. doi: 10.1002/cpz1.70010. Curr Protoc. 2024. PMID: 39499075 Free PMC article.
-
Profiling the genome-wide landscape of tandem repeat expansions.Nucleic Acids Res. 2019 Sep 5;47(15):e90. doi: 10.1093/nar/gkz501. Nucleic Acids Res. 2019. PMID: 31194863 Free PMC article.
-
A comparison of software for analysis of rare and common short tandem repeat (STR) variation using human genome sequences from clinical and population-based samples.PLoS One. 2024 Apr 1;19(4):e0300545. doi: 10.1371/journal.pone.0300545. eCollection 2024. PLoS One. 2024. PMID: 38558075 Free PMC article.
-
Recent advances in the detection of repeat expansions with short-read next-generation sequencing.F1000Res. 2018 Jun 13;7:F1000 Faculty Rev-736. doi: 10.12688/f1000research.13980.1. eCollection 2018. F1000Res. 2018. PMID: 29946432 Free PMC article. Review.
-
Advancing genomic technologies and clinical awareness accelerates discovery of disease-associated tandem repeat sequences.Genome Res. 2022 Jan;32(1):1-27. doi: 10.1101/gr.269530.120. Epub 2021 Dec 29. Genome Res. 2022. PMID: 34965938 Free PMC article. Review.
References
-
- Brais B. Correction: Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998. Aug;19(4):404–404. - PubMed
-
- Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, et al. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain. 2019. Nov 1;142(11):3360–6. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous