

This is a preprint.
Orientia tsutsugamushi: analysis of the mobilome of a highly fragmented and repetitive genome reveals ongoing lateral gene transfer in an obligate intracellular bacterium
- PMID: 37215039
- PMCID: PMC10197636
- DOI: 10.1101/2023.05.11.540415
Orientia tsutsugamushi: analysis of the mobilome of a highly fragmented and repetitive genome reveals ongoing lateral gene transfer in an obligate intracellular bacterium
Update in
-
Orientia tsutsugamushi: comprehensive analysis of the mobilome of a highly fragmented and repetitive genome reveals the capacity for ongoing lateral gene transfer in an obligate intracellular bacterium.mSphere. 2023 Dec 20;8(6):e0026823. doi: 10.1128/msphere.00268-23. Epub 2023 Oct 18. mSphere. 2023. PMID: 37850800 Free PMC article.
Abstract
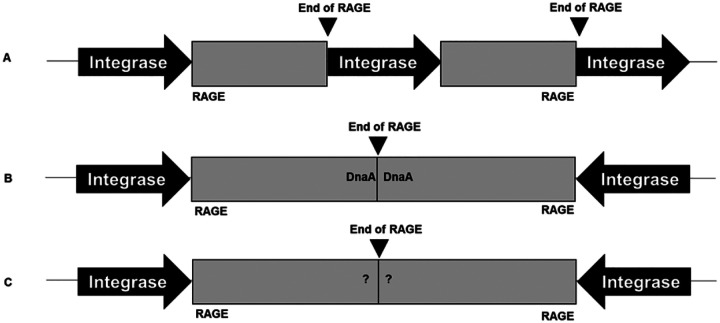
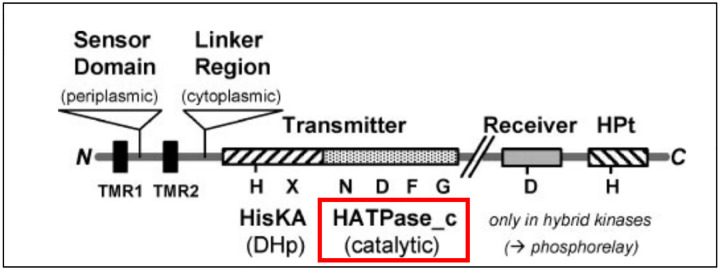
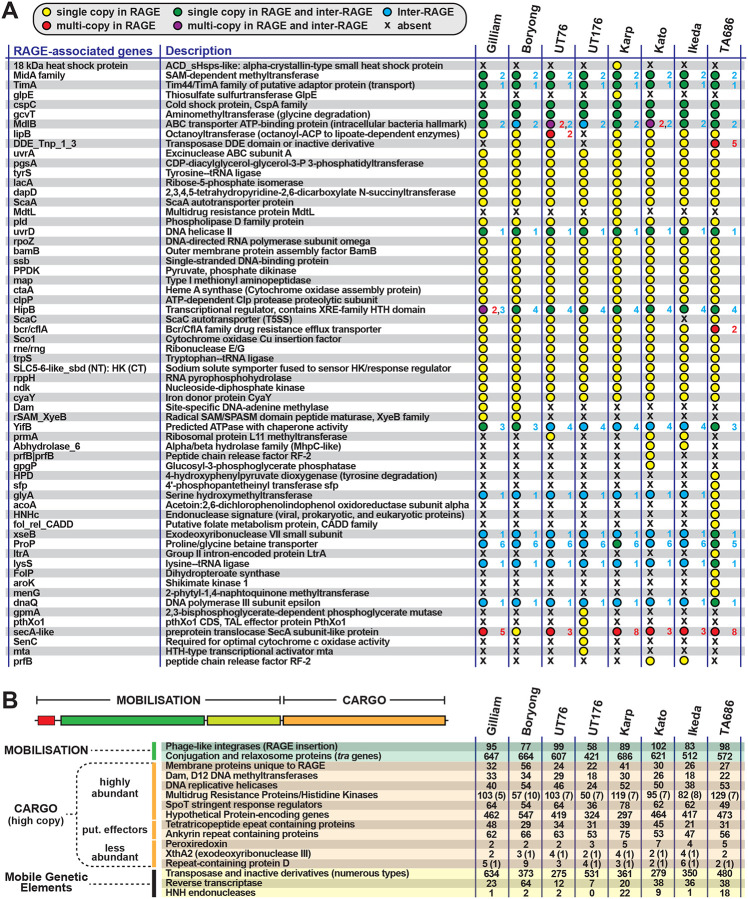
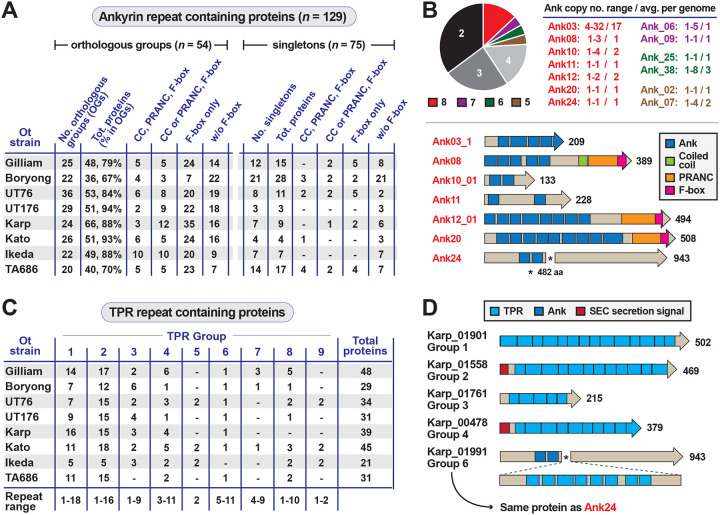
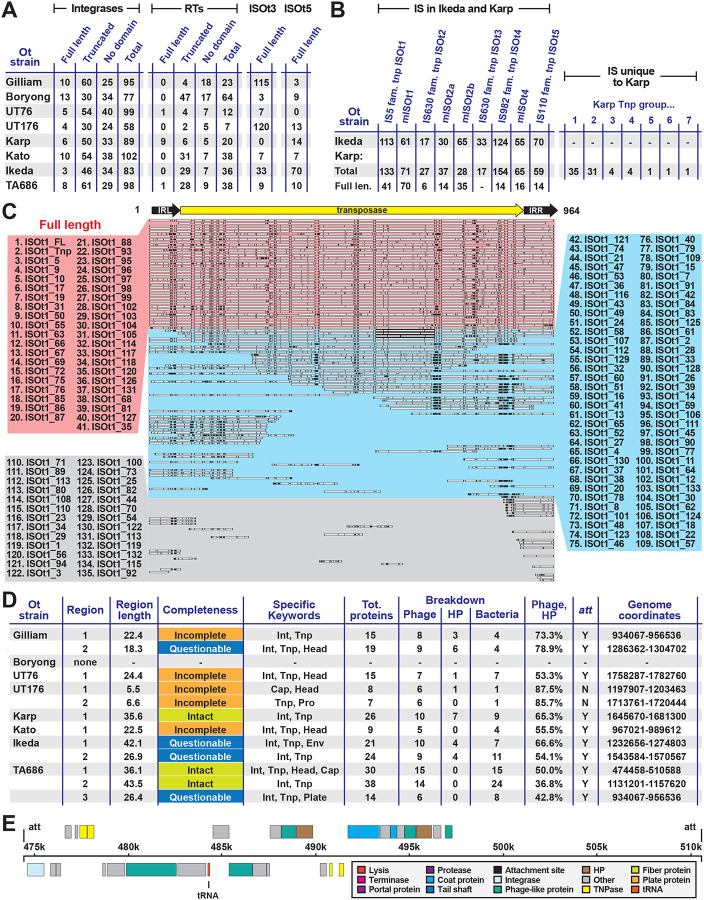
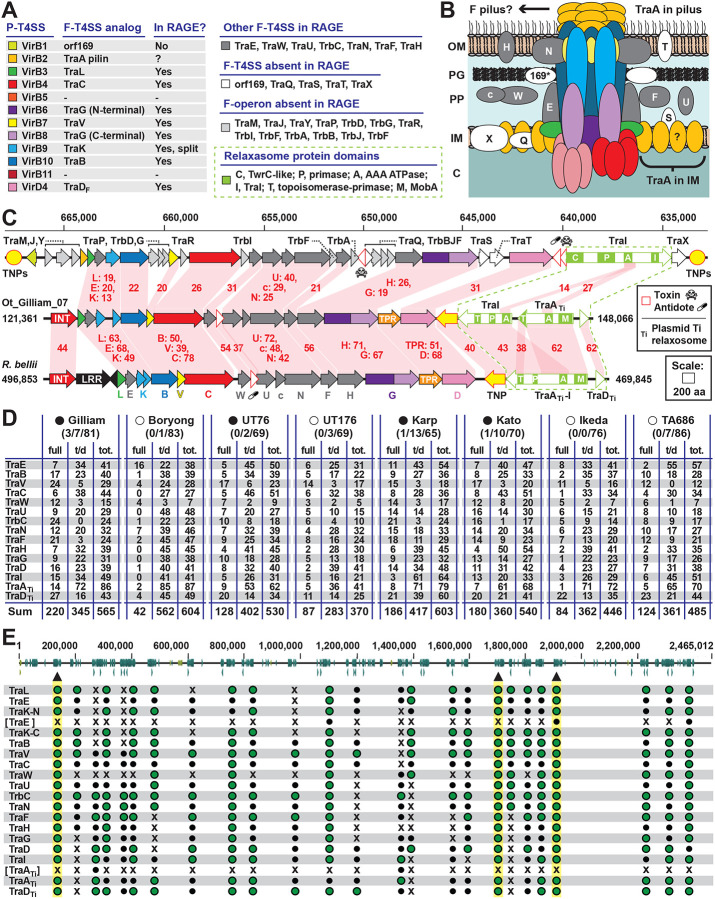
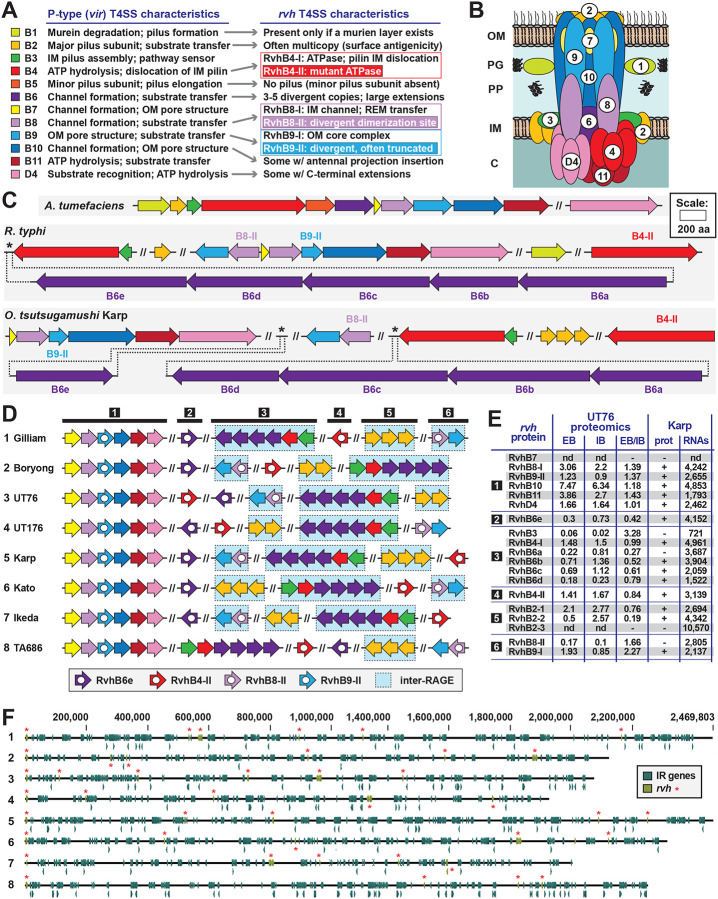
The rickettsial human pathogen Orientia tsutsugamushi (Ot) is an obligate intracellular Gram-negative bacterium with one of the most highly fragmented and repetitive genomes of any organism. Around 50% of its ~2.3 Mb genome is comprised of repetitive DNA that is derived from the highly proliferated Rickettsiales amplified genetic element (RAGE). RAGE is an integrative and conjugative element (ICE) that is present in a single Ot genome in up to 92 copies, most of which are partially or heavily degraded. In this report, we analysed RAGEs in eight fully sequenced Ot genomes and manually curated and reannotated all RAGE-associated genes, including those encoding DNA mobilisation proteins, P-type (vir) and F-type (tra) type IV secretion system (T4SS) components, Ankyrin repeat- and tetratricopeptide repeat-containing effectors, and other piggybacking cargo. Originally, the heavily degraded Ot RAGEs led to speculation that they are remnants of historical ICEs that are no longer active. Our analysis, however, identified two Ot genomes harbouring one or more intact RAGEs with complete F-T4SS genes essential for mediating ICE DNA transfer. As similar ICEs have been identified in unrelated rickettsial species, we assert that RAGEs play an ongoing role in lateral gene transfer within the Rickettsiales. Remarkably, we also identified in several Ot genomes remnants of prophages with no similarity to other rickettsial prophages. Together these findings indicate that, despite their obligate intracellular lifestyle and host range restricted to mites, rodents and humans, Ot genomes are highly dynamic and shaped through ongoing invasions by mobile genetic elements and viruses.
Keywords: Orientia tsutsugamushi; Rickettsiales; bacteriophage; comparative genomics; integrative and conjugative elements; intracellular pathogens; lateral gene transfer; mobile genetic elements; obligate intracellular bacteria.
Conflict of interest statement
Conflict of Interest The authors declare no conflict of interest.
Figures



Similar articles
-
Orientia tsutsugamushi: comprehensive analysis of the mobilome of a highly fragmented and repetitive genome reveals the capacity for ongoing lateral gene transfer in an obligate intracellular bacterium.mSphere. 2023 Dec 20;8(6):e0026823. doi: 10.1128/msphere.00268-23. Epub 2023 Oct 18. mSphere. 2023. PMID: 37850800 Free PMC article.
-
A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle.J Bacteriol. 2012 Jan;194(2):376-94. doi: 10.1128/JB.06244-11. Epub 2011 Nov 4. J Bacteriol. 2012. PMID: 22056929 Free PMC article.
-
The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes.Proc Natl Acad Sci U S A. 2007 May 8;104(19):7981-6. doi: 10.1073/pnas.0611553104. Epub 2007 May 2. Proc Natl Acad Sci U S A. 2007. PMID: 17483455 Free PMC article.
-
Rickettsial evolution in the light of comparative genomics.Biol Rev Camb Philos Soc. 2011 May;86(2):379-405. doi: 10.1111/j.1469-185X.2010.00151.x. Epub 2010 Aug 17. Biol Rev Camb Philos Soc. 2011. PMID: 20716256 Review.
-
New perspectives on rickettsial evolution from new genome sequences of rickettsia, particularly R. canadensis, and Orientia tsutsugamushi.Ann N Y Acad Sci. 2005 Dec;1063:47-63. doi: 10.1196/annals.1355.006. Ann N Y Acad Sci. 2005. PMID: 16481489 Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous