

Canonical correlation analysis for multi-omics: Application to cross-cohort analysis
- PMID: 37216410
- PMCID: PMC10237647
- DOI: 10.1371/journal.pgen.1010517
Canonical correlation analysis for multi-omics: Application to cross-cohort analysis
Abstract
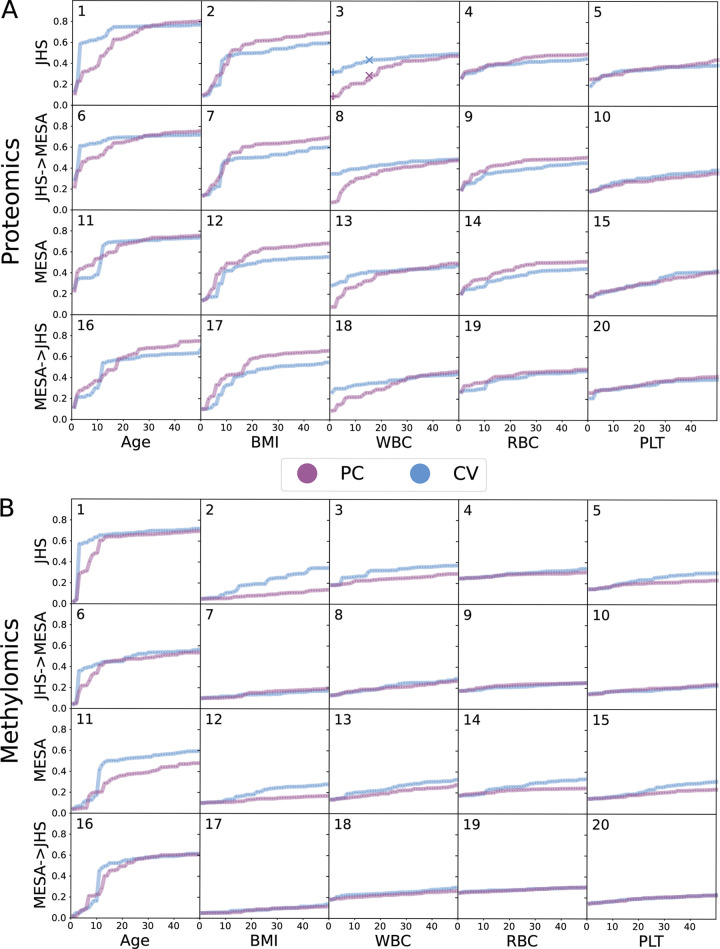
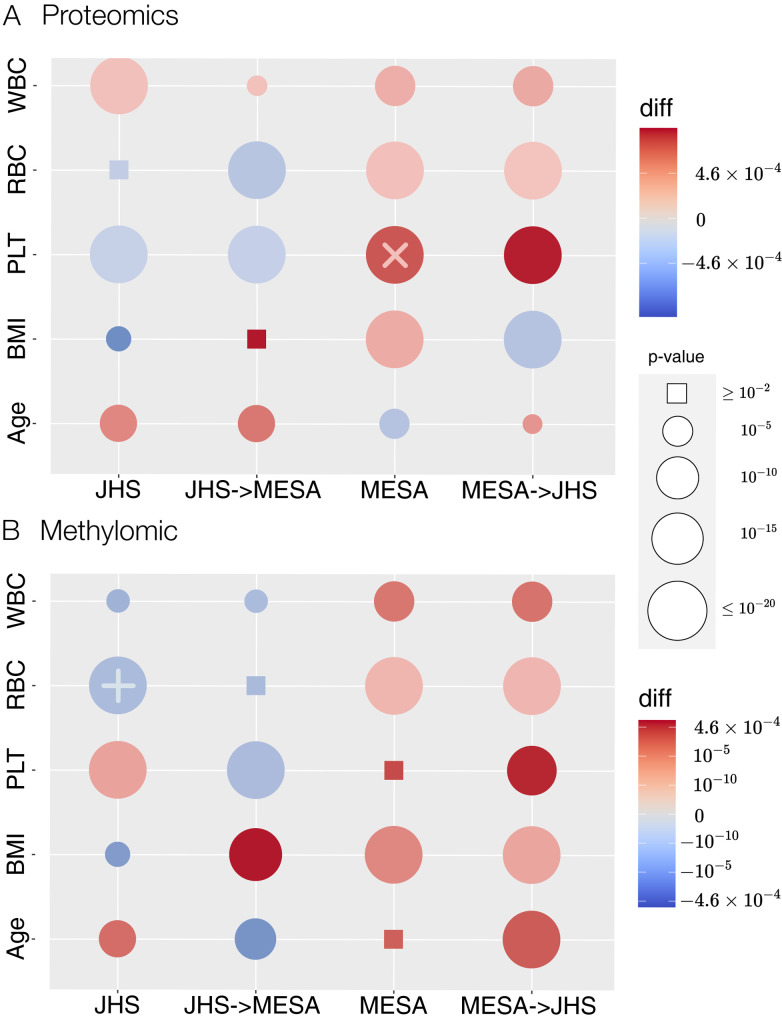
Integrative approaches that simultaneously model multi-omics data have gained increasing popularity because they provide holistic system biology views of multiple or all components in a biological system of interest. Canonical correlation analysis (CCA) is a correlation-based integrative method designed to extract latent features shared between multiple assays by finding the linear combinations of features-referred to as canonical variables (CVs)-within each assay that achieve maximal across-assay correlation. Although widely acknowledged as a powerful approach for multi-omics data, CCA has not been systematically applied to multi-omics data in large cohort studies, which has only recently become available. Here, we adapted sparse multiple CCA (SMCCA), a widely-used derivative of CCA, to proteomics and methylomics data from the Multi-Ethnic Study of Atherosclerosis (MESA) and Jackson Heart Study (JHS). To tackle challenges encountered when applying SMCCA to MESA and JHS, our adaptations include the incorporation of the Gram-Schmidt (GS) algorithm with SMCCA to improve orthogonality among CVs, and the development of Sparse Supervised Multiple CCA (SSMCCA) to allow supervised integration analysis for more than two assays. Effective application of SMCCA to the two real datasets reveals important findings. Applying our SMCCA-GS to MESA and JHS, we identified strong associations between blood cell counts and protein abundance, suggesting that adjustment of blood cell composition should be considered in protein-based association studies. Importantly, CVs obtained from two independent cohorts also demonstrate transferability across the cohorts. For example, proteomic CVs learned from JHS, when transferred to MESA, explain similar amounts of blood cell count phenotypic variance in MESA, explaining 39.0% ~ 50.0% variation in JHS and 38.9% ~ 49.1% in MESA. Similar transferability was observed for other omics-CV-trait pairs. This suggests that biologically meaningful and cohort-agnostic variation is captured by CVs. We anticipate that applying our SMCCA-GS and SSMCCA on various cohorts would help identify cohort-agnostic biologically meaningful relationships between multi-omics data and phenotypic traits.
Copyright: © 2023 Jiang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: LMR is a consultant for the TOPMed Administrative Coordinating Center (through Westat).
Figures



References
Publication types
MeSH terms
Grants and funding
- HHSN268201100037C/HL/NHLBI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- HHSN268201600032C/ES/NIEHS NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- R01 HL105756/HL/NHLBI NIH HHS/United States
- T32 HL129982/HL/NHLBI NIH HHS/United States
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- N01 HC095166/HL/NHLBI NIH HHS/United States
- N01 HC095160/HL/NHLBI NIH HHS/United States
- 75N92020D00002/HL/NHLBI NIH HHS/United States
- HHSN268201500003C/HL/NHLBI NIH HHS/United States
- N01 HC095161/HL/NHLBI NIH HHS/United States
- 75N92020D00005/HL/NHLBI NIH HHS/United States
- N01 HC095168/HL/NHLBI NIH HHS/United States
- R01 HL120393/HL/NHLBI NIH HHS/United States
- UL1 TR001079/TR/NCATS NIH HHS/United States
- N02 HL064278/HL/NHLBI NIH HHS/United States
- N01 HC095169/HL/NHLBI NIH HHS/United States
- R01 AG075884/AG/NIA NIH HHS/United States
- 75N92020D00001/HL/NHLBI NIH HHS/United States
- HHSN268201300048C/HL/NHLBI NIH HHS/United States
- N01 HC095167/HL/NHLBI NIH HHS/United States
- N01 HC095159/HL/NHLBI NIH HHS/United States
- 75N92020D00003/HL/NHLBI NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- HHSN268201300049C/HL/NHLBI NIH HHS/United States
- HHSN268201300047C/HL/NHLBI NIH HHS/United States
- UL1 TR001420/TR/NCATS NIH HHS/United States
- 75N92020D00004/HL/NHLBI NIH HHS/United States
- HHSN268201300050C/HL/NHLBI NIH HHS/United States
- N01 HC095163/HL/NHLBI NIH HHS/United States
- 75N92020D00007/HL/NHLBI NIH HHS/United States
- R01 HL146500/HL/NHLBI NIH HHS/United States
- HHSN268201500003I/HL/NHLBI NIH HHS/United States
- KL2 TR002490/TR/NCATS NIH HHS/United States
- UL1 TR000040/TR/NCATS NIH HHS/United States
- HHSN268201300046C/HL/NHLBI NIH HHS/United States
- 75N92020D00006/HL/NHLBI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- N01 HC095162/HL/NHLBI NIH HHS/United States
- UL1 TR001881/TR/NCATS NIH HHS/United States
- N01 HC095165/HL/NHLBI NIH HHS/United States
- N01 HC095164/HL/NHLBI NIH HHS/United States
