PROTAC-Induced Glycogen Synthase Kinase 3β Degradation as a Potential Therapeutic Strategy for Alzheimer's Disease
- PMID: 37218653
- PMCID: PMC10251479
- DOI: 10.1021/acschemneuro.3c00096
PROTAC-Induced Glycogen Synthase Kinase 3β Degradation as a Potential Therapeutic Strategy for Alzheimer's Disease
Abstract
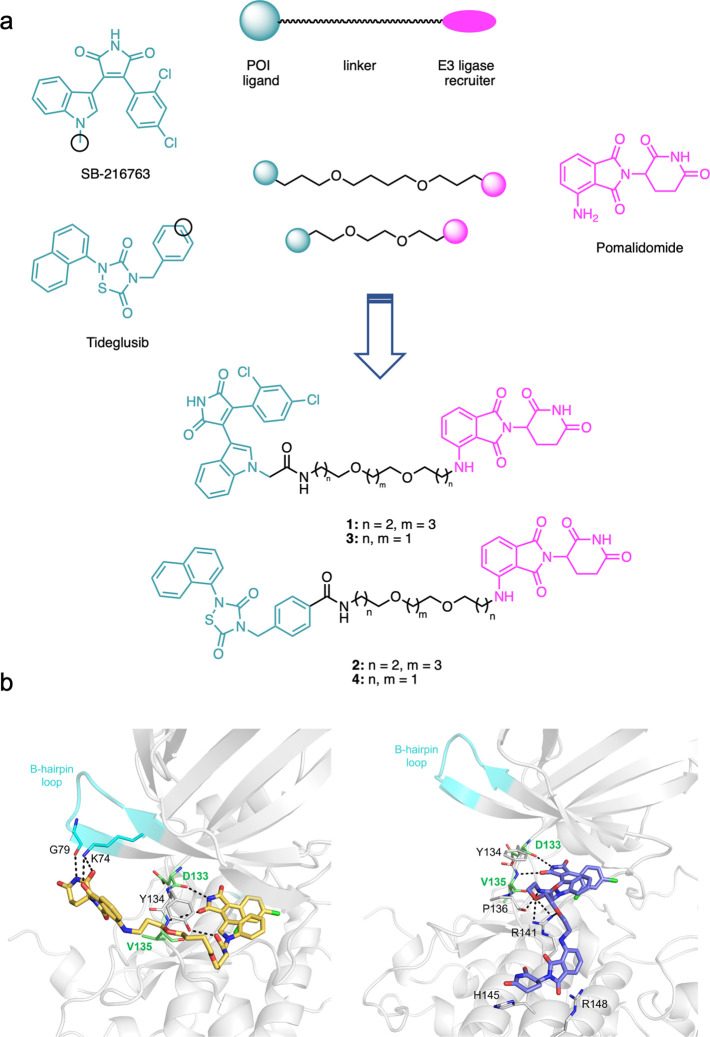
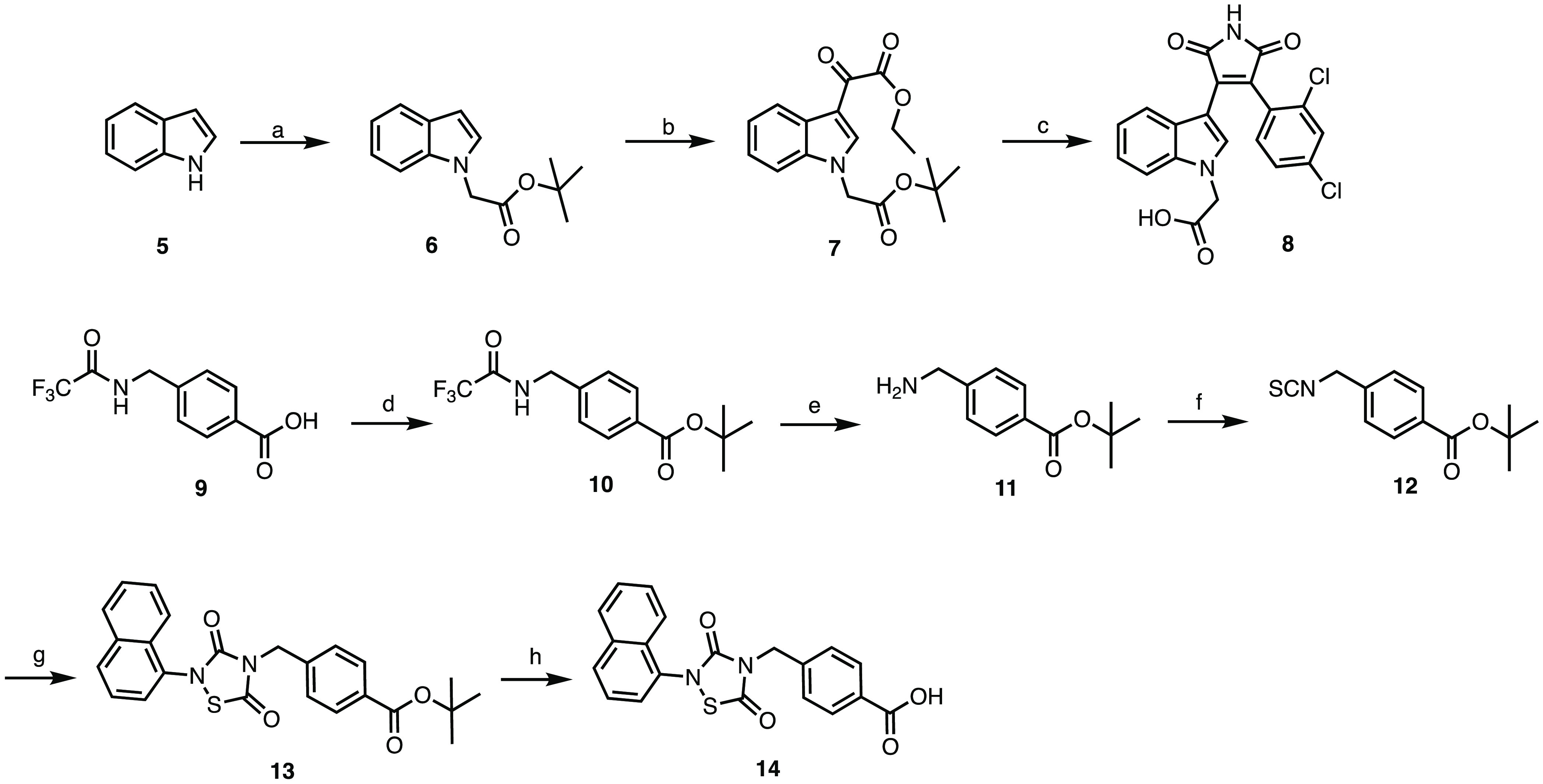
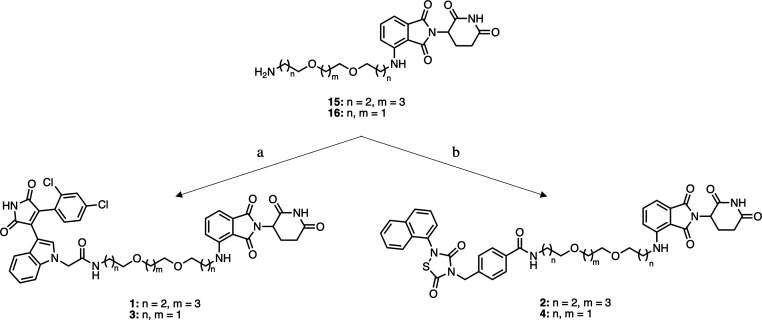
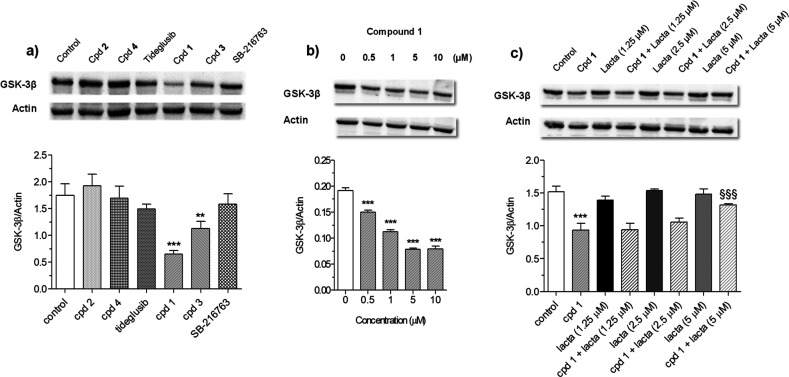
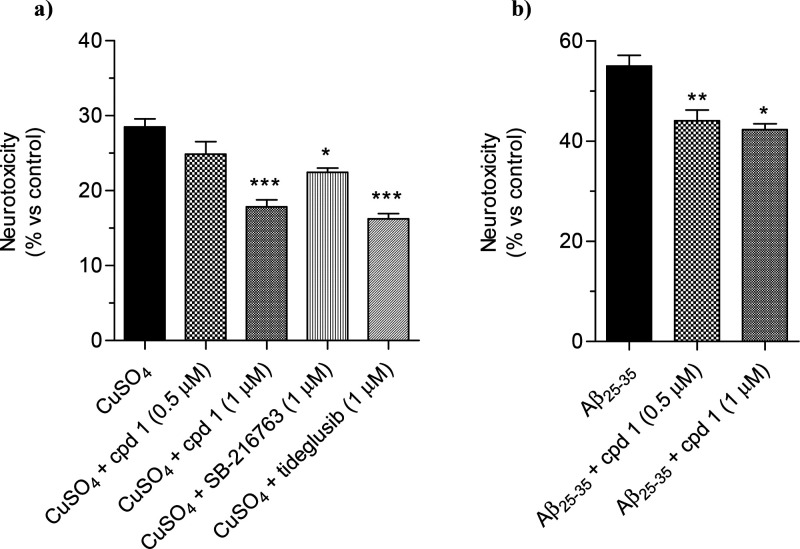
Glycogen synthase kinase 3β (GSK-3β) is a serine/threonine kinase and an attractive therapeutic target for Alzheimer's disease. Based on proteolysis-targeting chimera (PROTAC) technology, a small set of novel GSK-3β degraders was designed and synthesized by linking two different GSK-3β inhibitors, SB-216763 and tideglusib, to pomalidomide, as E3 recruiting element, through linkers of different lengths. Compound 1 emerged as the most effective PROTAC being nontoxic up to 20 μM to neuronal cells and already able to degrade GSK-3β starting from 0.5 μM in a dose-dependent manner. PROTAC 1 significantly reduced the neurotoxicity induced by Aβ25-35 peptide and CuSO4 in SH-SY5Y cells in a dose-dependent manner. Based on its encouraging features, PROTAC 1 may serve as a starting point to develop new GSK-3β degraders as potential therapeutic agents.
Keywords: Alzheimer’s disease; chemical knockdown; glycogen synthase kinase 3β; protein degradation; proteolysis targeting chimeras.
Conflict of interest statement
The authors declare no competing financial interest.
Figures