

[Molecular therapies: present and future in neuromuscular diseases]
- PMID: 37221259
- PMCID: PMC10204661
- DOI: 10.1007/s00115-023-01495-3
[Molecular therapies: present and future in neuromuscular diseases]
Abstract
Background: The possibilities in the field of molecular therapies of neuromuscular diseases have rapidly developed in recent years. First compounds are already available in clinical practice and numerous other substances are in advanced phases of clinical trials. This article gives an exemplary overview of the current state of clinical research in molecular therapies of neuromuscular diseases. It also gives a view into the near future of the clinical application, including the challenges.
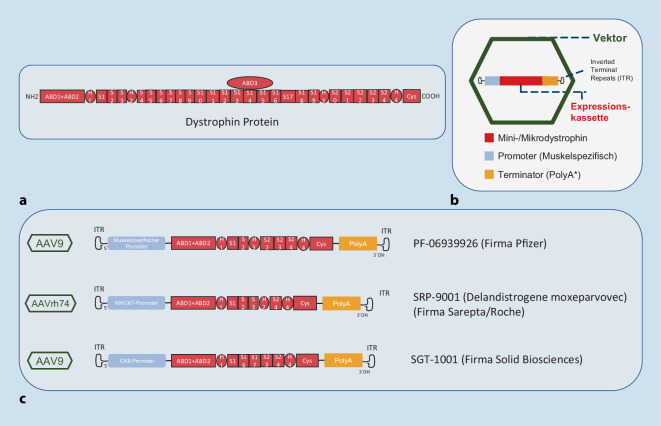
Discussion: Using Duchenne muscular dystrophy (DMD) and myotubular myopathy as examples, the principles of gene addition in monogenetic skeletal muscle diseases, which are already manifested in childhood are described. In addition to initial successes, the challenges and setbacks hindering the approval and regular clinical application of further compounds are demonstrated. Furthermore, the state of current clinical research in Becker-Kiener muscular dystrophy (BMD) and the numerous forms of limb-girdle muscular dystrophy (LGMD) are summarized. Numerous new therapeutic approaches and a corresponding outlook are also shown for facioscapulohumeral muscular dystrophy (FSHD), Pompe disease, and myotonic dystrophy.
Conclusion: Clinical research in the field of molecular therapy of neuromuscular diseases is one of the pacesetters of modern precision medicine; however, challenges need to be seen, jointly addressed and overcome in the future.
Zusammenfassung: HINTERGRUND: Die Möglichkeiten im Bereich der molekularen Therapien neuromuskulärer Erkrankungen haben sich in den letzten Jahren rasant entwickelt. Erste Präparate stehen im klinischen Alltag zur Verfügung, zahlreiche weitere Substanzen befinden sich in fortgeschrittenen Phasen der klinischen Prüfung. Dieser Beitrag gibt einen aktuellen, exemplarischen Überblick über den Stand der klinischen Forschung im Bereich der molekularen Therapien neuromuskulärer Erkrankungen mit Blick in die nahe Zukunft der klinischen Anwendung inklusive deren Herausforderungen.
Diskussion: Am Beispiel der Duchenne Muskeldystrophie und der myotubulären Myopathie werden die Prinzipien der Genaddition bei monogenetischen Erkrankungen der Muskulatur beschrieben, die sich bereits im Kindesalter manifestieren. Neben ersten Erfolgen werden auch die Herausforderungen und Rückschläge aufgezeigt, die für eine Zulassung und klinische Regelanwendung überwunden werden müssen. Darüber hinaus fassen wir den Stand der klinischen Forschung im Bereich der Muskeldystrophie Typ Becker-Kiener und in Auswahl für die Gliedergürteldystrophien zusammen. Zahlreiche neue therapeutische Ansätze und ein entsprechender Ausblick werden auch für die fazioskapulohumerale Muskeldystrophie (FSHD), den Morbus Pompe und die myotone Dystrophie gezeigt.
Schlussfolgerung: Die klinische Forschung im Bereich der molekularen Therapien neuromuskulärer Erkrankungen gehört zu den Schrittmachern der modernen Präzisionsmedizin. Herausforderungen müssen aber gesehen und in Zukunft gemeinsam adressiert und überwunden werden.
Keywords: Becker muscular dystrophy; Duchenne muscular dystrophy; Muscular dystrophy, facioscapulohumeral; Muscular dystrophy, limb-girdle; Pompe disease.
© 2023. The Author(s), under exclusive licence to Springer Medizin Verlag GmbH, ein Teil von Springer Nature.
Similar articles
-
Perturbation of muscle metabolism in patients with muscular dystrophy in early or acute phase of disease: In vitro, high resolution NMR spectroscopy based analysis.Clin Chim Acta. 2018 Mar;478:171-181. doi: 10.1016/j.cca.2017.12.036. Epub 2017 Dec 24. Clin Chim Acta. 2018. PMID: 29278724
-
The Muscular Dystrophy Association's neuroMuscular ObserVational Research Data Hub (MOVR): Design, Methods, and Initial Observations.J Neuromuscul Dis. 2023;10(3):365-380. doi: 10.3233/JND-221551. J Neuromuscul Dis. 2023. PMID: 36911943 Free PMC article.
-
[Innovative therapeutic approaches for hereditary neuromuscular diseases].Nervenarzt. 2018 Oct;89(10):1115-1122. doi: 10.1007/s00115-018-0599-9. Nervenarzt. 2018. PMID: 30171303 Review. German.
-
Abnormal lipid metabolism in skeletal muscle tissue of patients with muscular dystrophy: In vitro, high-resolution NMR spectroscopy based observation in early phase of the disease.Magn Reson Imaging. 2017 May;38:163-173. doi: 10.1016/j.mri.2017.01.001. Epub 2017 Jan 7. Magn Reson Imaging. 2017. PMID: 28069416
-
New pharmacotherapies for genetic neuromuscular disorders: opportunities and challenges.Expert Rev Clin Pharmacol. 2019 Aug;12(8):757-770. doi: 10.1080/17512433.2019.1634543. Epub 2019 Jul 2. Expert Rev Clin Pharmacol. 2019. PMID: 31220956 Review.
References
-
- Angelini C. Calpainopathy. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(®) Seattle: University of Washington; 1993. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
