

Sting and p53 DNA repair pathways are compromised in Alzheimer's disease
- PMID: 37221295
- PMCID: PMC10206146
- DOI: 10.1038/s41598-023-35533-6
Sting and p53 DNA repair pathways are compromised in Alzheimer's disease
Abstract
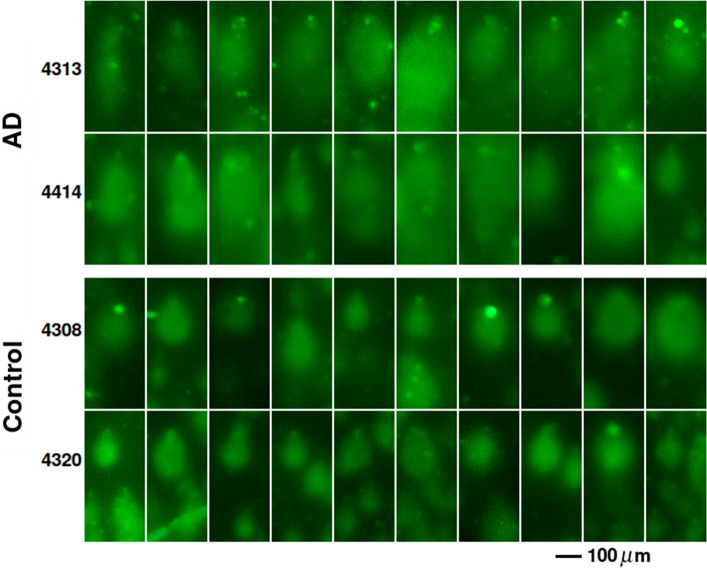
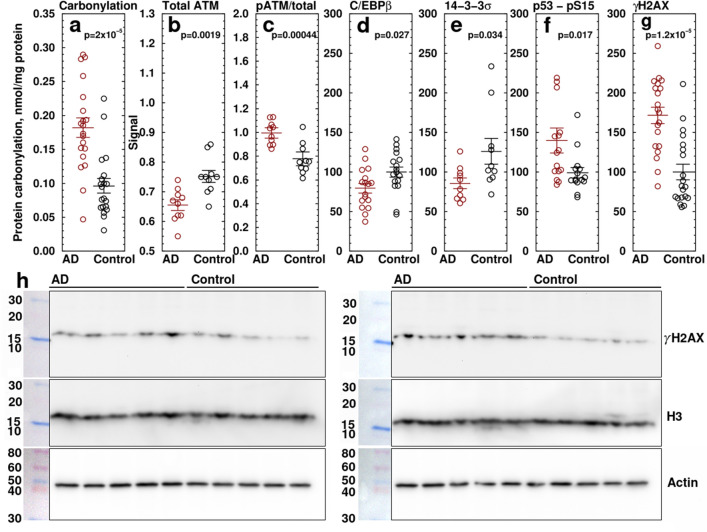
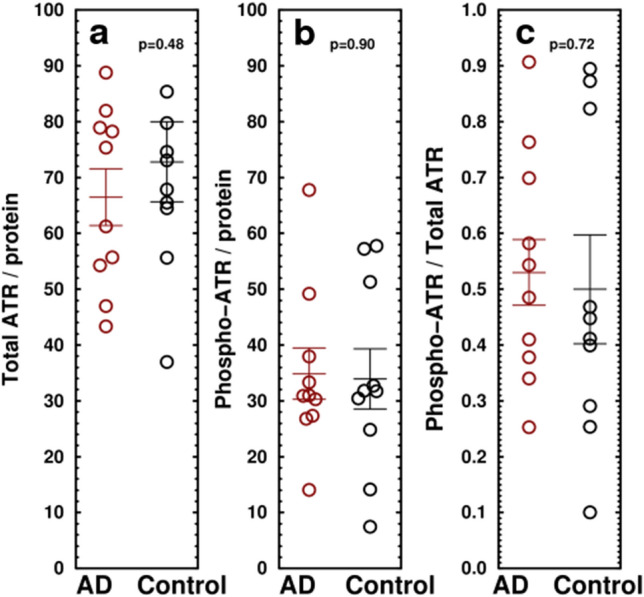
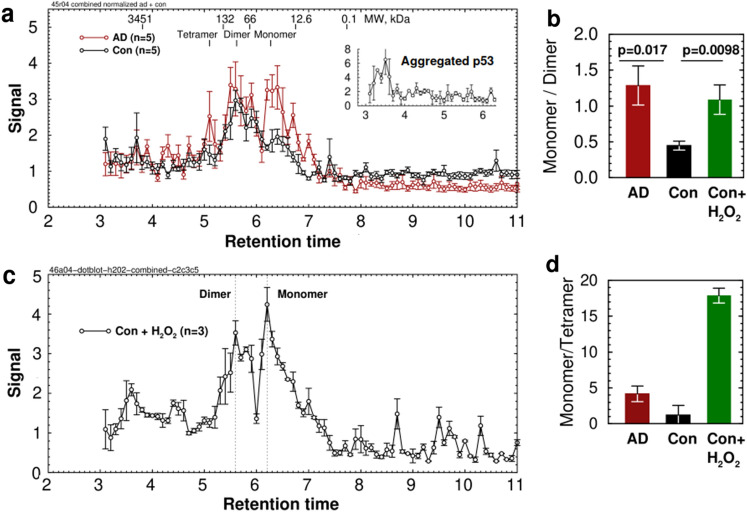
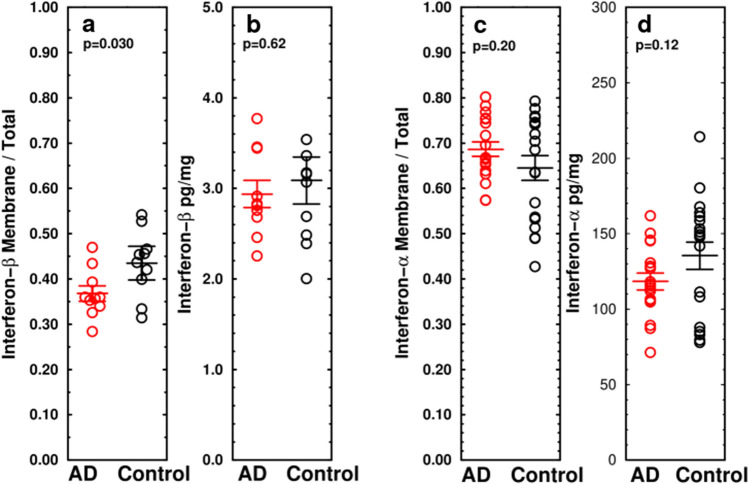
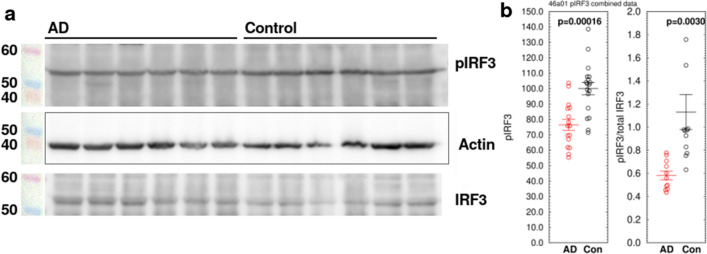
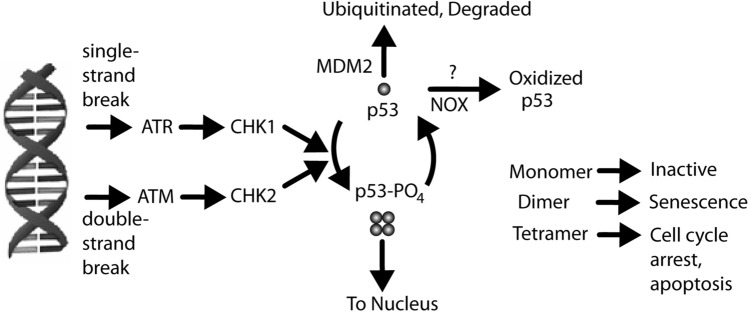
Alzheimer's disease (AD) is the most common cause of dementia. A common finding in AD is DNA damage. Double-strand DNA breaks (DSBs) are particularly hazardous to neurons because their post-mitotic state forces neurons to rely on error-prone and potentially mutagenic mechanisms to repair DNA breaks. However, it remains unclear whether DNA damage results from increased DNA damage or failure of DNA repair. Oligomerization of the tumor suppressor protein p53 is an essential part of DSB repair, and p53 phosphorylated on S15 is an indicator of DNA damage. We report that the monomer:dimer ratio of phosphorylated (S15) p53 is increased by 2.86-fold in temporal lobes of AD patients compared to age-matched controls, indicating that p53 oligomerization is compromised in AD. In vitro oxidation of p53 with 100 nM H2O2 produced a similar shift in the monomer:dimer ratio. A COMET test showed a higher level of DNA degradation in AD consistent with double-strand DNA damage or inhibition of repair. Protein carbonylation was also elevated (190% of control), indicating elevated oxidative stress in AD patients. Levels of the DNA repair support protein 14-3-3σ, γ-H2AX, a phosphorylated histone marking double strand DNA breaks, and phosphorylated ataxia telangiectasia mutated (ATM) protein were all increased. cGAS-STING-interferon signaling was impaired in AD and was accompanied by a depletion of STING protein from Golgi and a failure to elevate interferon despite the presence of DSBs. The results suggest that oxidation of p53 by ROS could inhibit the DDR and decrease its ability to orchestrate DSB repair by altering the oligomerization state of p53. The failure of immune-stimulated DNA repair may contribute to cell loss in AD and suggests new therapeutic targets for AD.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Relationship between DNA double-strand break rejoining and cell survival after exposure to ionizing radiation in human fibroblast strains with differing ATM/p53 status: implications for evaluation of clinical radiosensitivity.Int J Radiat Oncol Biol Phys. 2006 Dec 1;66(5):1498-505. doi: 10.1016/j.ijrobp.2006.08.064. Int J Radiat Oncol Biol Phys. 2006. PMID: 17126209
-
Dynamic monitoring of oxidative DNA double-strand break and repair in cardiomyocytes.Cardiovasc Pathol. 2016 Mar-Apr;25(2):93-100. doi: 10.1016/j.carpath.2015.10.010. Epub 2015 Oct 31. Cardiovasc Pathol. 2016. PMID: 26764141 Free PMC article.
-
Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability.J Cell Sci. 2012 Jan 1;125(Pt 1):189-99. doi: 10.1242/jcs.092262. Epub 2012 Jan 20. J Cell Sci. 2012. PMID: 22266907
-
The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax.DNA Repair (Amst). 2010 Dec 10;9(12):1273-82. doi: 10.1016/j.dnarep.2010.09.013. Epub 2010 Oct 30. DNA Repair (Amst). 2010. PMID: 21036673 Review.
-
An Adaptive Role for DNA Double-Strand Breaks in Hippocampus-Dependent Learning and Memory.Int J Mol Sci. 2022 Jul 28;23(15):8352. doi: 10.3390/ijms23158352. Int J Mol Sci. 2022. PMID: 35955487 Free PMC article. Review.
Cited by
-
Inflammation and DNA methylation in Alzheimer's disease: mechanisms of epigenetic remodelling by immune cell oxidants in the ageing brain.Redox Rep. 2024 Dec;29(1):2428152. doi: 10.1080/13510002.2024.2428152. Epub 2024 Nov 23. Redox Rep. 2024. PMID: 39579010 Free PMC article. Review.
-
Inhibition of the cGAS-STING pathway via an endogenous copper ion-responsive covalent organic framework nanozyme for Alzheimer's disease treatment.Chem Sci. 2025 Mar 11;16(17):7215-7226. doi: 10.1039/d4sc07963a. eCollection 2025 Apr 30. Chem Sci. 2025. PMID: 40144496 Free PMC article.
-
Insights into targeted ferroptosis in mechanisms, biology, and role of Alzheimer's disease: an update.Front Aging Neurosci. 2025 Jul 21;17:1587986. doi: 10.3389/fnagi.2025.1587986. eCollection 2025. Front Aging Neurosci. 2025. PMID: 40761698 Free PMC article. Review.
-
The Dual Role of Amyloid Beta-Peptide in Oxidative Stress and Inflammation: Unveiling Their Connections in Alzheimer's Disease Etiopathology.Antioxidants (Basel). 2024 Oct 8;13(10):1208. doi: 10.3390/antiox13101208. Antioxidants (Basel). 2024. PMID: 39456461 Free PMC article. Review.
-
The STING Signaling: A Novel Target for Central Nervous System Diseases.Cell Mol Neurobiol. 2025 Apr 7;45(1):33. doi: 10.1007/s10571-025-01550-4. Cell Mol Neurobiol. 2025. PMID: 40195137 Free PMC article. Review.
References
-
- Vemuri P, Knopman DS, Lesnick TG, Przybelski SA, Mielke MM, Graff-Radford J, Murray ME, Roberts RO, Vassilaki M, Lowe VJ, Machulda MM, Jones DT, Petersen RC, Jack CR., Jr Evaluation of amyloid protective factors and Alzheimer disease neurodegeneration protective factors in elderly individuals. JAMA Neurol. 2017;74(6):718–726. doi: 10.1001/jamaneurol.2017.0244. - DOI - PMC - PubMed
-
- Soheili-Nezhad S, van der Linden RJ, Olde Rikkert M, Sprooten E, Poelmans G. Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer's disease: Implications for disease etiology. Alzheimer's Dementia. 2021;17(3):489–499. doi: 10.1002/alz.12211. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
