

A novel 1p13.2 deletion associates with neurodevelopmental disorders in a three-generation pedigree
- PMID: 37221554
- PMCID: PMC10207759
- DOI: 10.1186/s12920-023-01534-7
A novel 1p13.2 deletion associates with neurodevelopmental disorders in a three-generation pedigree
Abstract
Background: A multitude of studies have highlighted that copy number variants (CNVs) are associated with neurodevelopmental disorders (NDDs) characterized by a wide range of clinical characteristics. Benefiting from CNV calling from WES data, WES has emerged as a more powerful and cost-effective molecular diagnostic tool, which has been widely used for the diagnosis of genetic diseases, especially NDDs. To our knowledge, isolated deletions on chromosome 1p13.2 are rare. To date, only a few patients were reported with 1p13.2 deletions and most of them were sporadic. Besides, the correlation between 1p13.2 deletions and NDDs remained unclear.
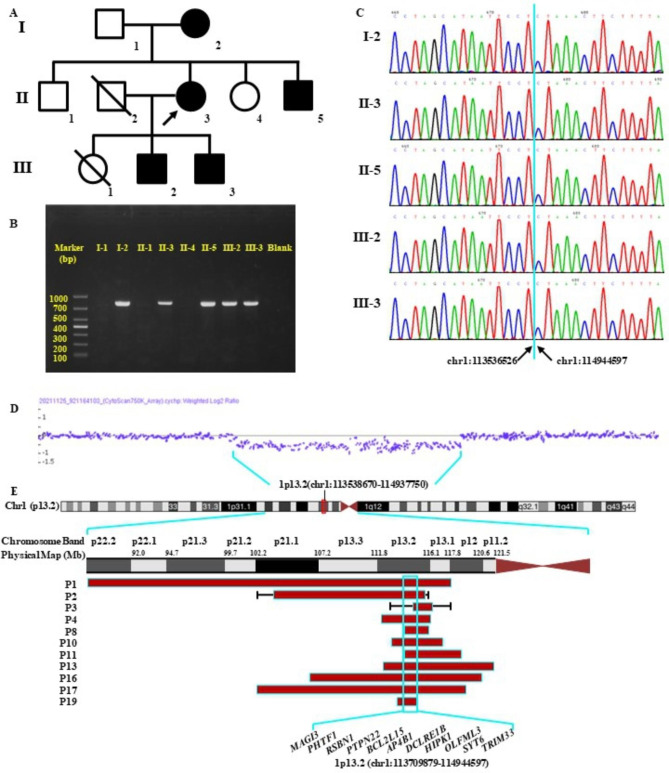
Case presentation: Here, we first reported five members in a three-generation Chinese family who presented with NDDs and carried a novel 1.41 Mb heterozygous 1p13.2 deletion with precise breakpoints. The diagnostic deletion contained 12 protein-coding genes and was observed to segregate with NDDs among the members of our reported family. Whether those genes contribute to the patient's phenotypes is still inconclusive.
Conclusions: We hypothesized that the NDD phenotype of our patients was caused by the diagnostic 1p13.2 deletion. However, further in-depth functional experiments are still needed to establish a 1p13.2 deletion-NDDs relationship. Our study might supplement the spectrum of 1p13.2 deletion-NDDs.
Keywords: 1p13.2 deletion; Molecular diagnosis; Neurodevelopmental disorders; Whole-exome sequencing.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
Similar articles
-
Incorporation of exome-based CNV analysis makes trio-WES a more powerful tool for clinical diagnosis in neurodevelopmental disorders: A retrospective study.Hum Mutat. 2021 Aug;42(8):990-1004. doi: 10.1002/humu.24222. Epub 2021 May 31. Hum Mutat. 2021. PMID: 34015165
-
A rigorous in silico genomic interrogation at 1p13.3 reveals 16 autosomal dominant candidate genes in syndromic neurodevelopmental disorders.Front Mol Neurosci. 2022 Oct 6;15:979061. doi: 10.3389/fnmol.2022.979061. eCollection 2022. Front Mol Neurosci. 2022. PMID: 36277487 Free PMC article.
-
Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders.Genome Med. 2021 Apr 19;13(1):63. doi: 10.1186/s13073-021-00870-6. Genome Med. 2021. PMID: 33874999 Free PMC article.
-
Familial 5.29 Mb deletion in chromosome Xq22.1-q22.3 with a normal phenotype: a rare pedigree and literature review.BMC Med Genomics. 2023 May 22;16(1):111. doi: 10.1186/s12920-023-01547-2. BMC Med Genomics. 2023. PMID: 37217926 Free PMC article. Review.
-
DNA Methylation Episignatures in Neurodevelopmental Disorders Associated with Large Structural Copy Number Variants: Clinical Implications.Int J Mol Sci. 2022 Jul 16;23(14):7862. doi: 10.3390/ijms23147862. Int J Mol Sci. 2022. PMID: 35887210 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
