

Genomic hallmarks and therapeutic implications of G0 cell cycle arrest in cancer
- PMID: 37221612
- PMCID: PMC10204193
- DOI: 10.1186/s13059-023-02963-4
Genomic hallmarks and therapeutic implications of G0 cell cycle arrest in cancer
Abstract
Background: Therapy resistance in cancer is often driven by a subpopulation of cells that are temporarily arrested in a non-proliferative G0 state, which is difficult to capture and whose mutational drivers remain largely unknown.
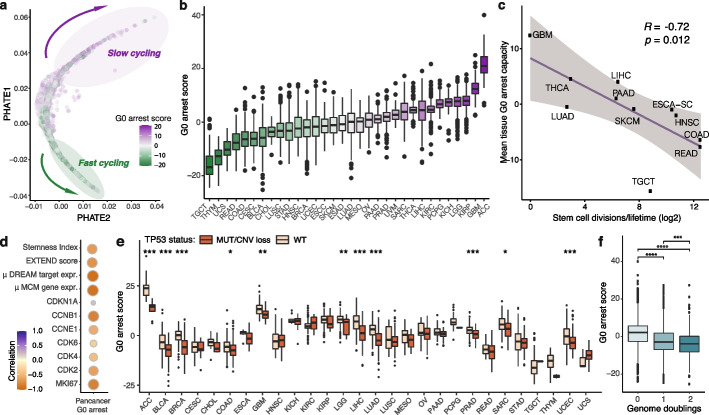
Results: We develop methodology to robustly identify this state from transcriptomic signals and characterise its prevalence and genomic constraints in solid primary tumours. We show that G0 arrest preferentially emerges in the context of more stable, less mutated genomes which maintain TP53 integrity and lack the hallmarks of DNA damage repair deficiency, while presenting increased APOBEC mutagenesis. We employ machine learning to uncover novel genomic dependencies of this process and validate the role of the centrosomal gene CEP89 as a modulator of proliferation and G0 arrest capacity. Lastly, we demonstrate that G0 arrest underlies unfavourable responses to various therapies exploiting cell cycle, kinase signalling and epigenetic mechanisms in single-cell data.
Conclusions: We propose a G0 arrest transcriptional signature that is linked with therapeutic resistance and can be used to further study and clinically track this state.
Keywords: Bulk/single-cell sequencing; Cancer; Cell cycle arrest; Data integration; G0; Genomic dependencies; Machine learning; Persister cells.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
Publication types
MeSH terms
Grants and funding
- C63833/A25729/CRUK_/Cancer Research UK/United Kingdom
- BB/R01356X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MR/T042184/1/MRC_/Medical Research Council/United Kingdom
- MC_UP_1605/8/MRC_/Medical Research Council/United Kingdom
- 25729/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
