

Temporal and coevolutionary analyses reveal the events driving the emergence and circulation of human mamastroviruses
- PMID: 37222427
- PMCID: PMC10262812
- DOI: 10.1080/22221751.2023.2217942
Temporal and coevolutionary analyses reveal the events driving the emergence and circulation of human mamastroviruses
Abstract
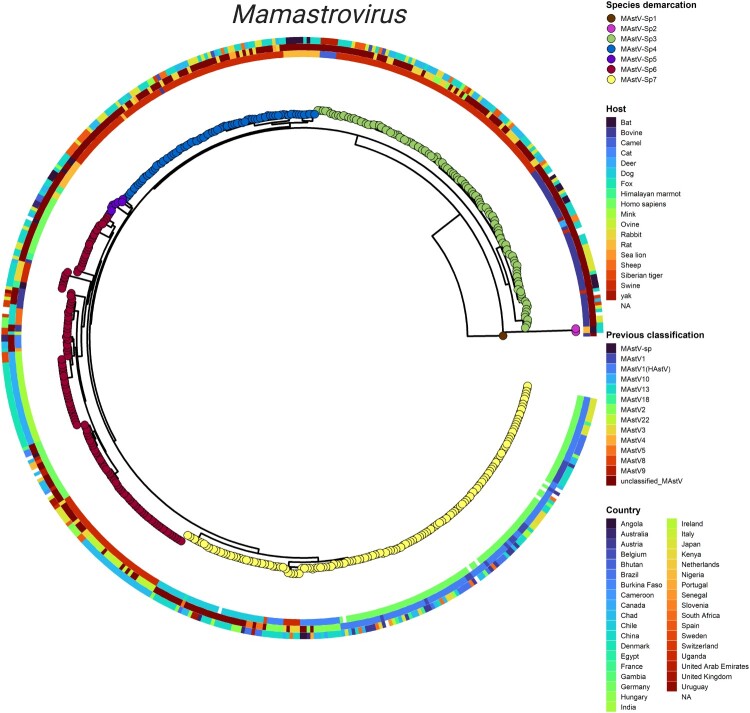
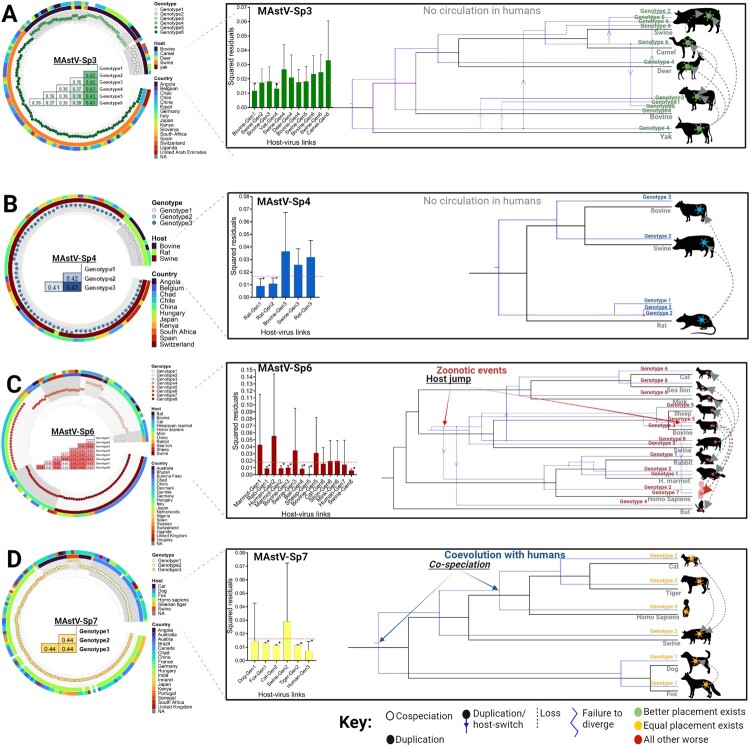
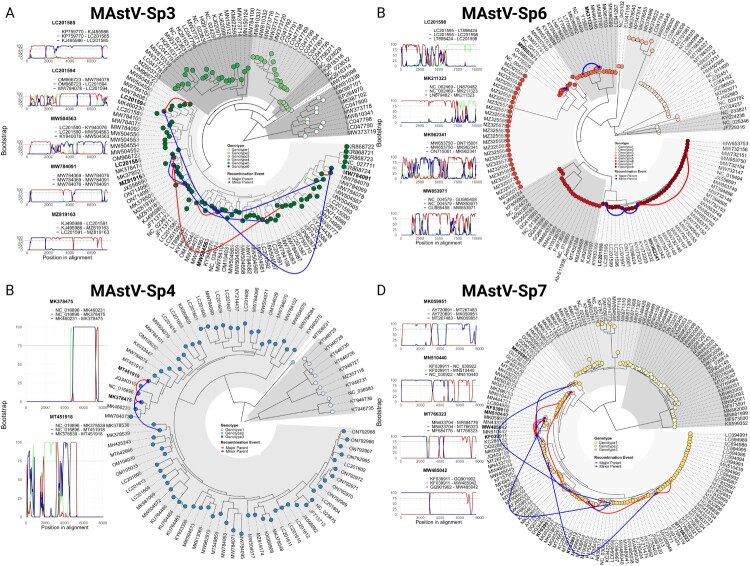
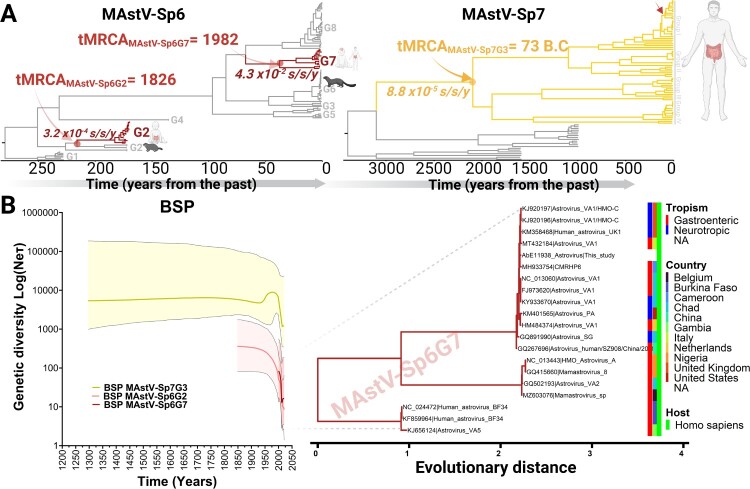
Characterized by high genetic diversity, broad host range, and resistance to adverse conditions, coupled with recent reports of neurotropic astroviruses circulating in humans, mamastroviruses pose a threat to public health. The current astrovirus classification system based on host source prevents determining whether strains with distinct tropism or virulence are emerging. By using integrated phylogeny, we propose a standardized demarcation of species and genotypes, with reproducible cut-off values that reconcile the pairwise sequence distribution, genetic distances between lineages, and the topological reconstruction of the Mamastrovirus genus. We further define the various links established by co-evolution and resolve the dynamics of transmission chains to identify host-jump events and the sources from which different mamastrovirus species circulating in humans have emerged. We observed that recombination is relatively infrequent and restricted to within genotypes. The well-known "human" astrovirus, defined here as mamastrovirus species 7, has co-speciated with humans, while there have been two additional host-jumps into humans from distinct hosts. Newly defined species 6 genotype 2, linked to severe gastroenteritis in children, resulted from a marmot to human jump taking place ∼200 years ago while species 6 genotype 7 (MastV-Sp6Gt7), linked to neurological disease in immunocompromised patients, jumped from bovines only ∼50 years ago. Through demographic reconstruction, we determined that the latter reached coalescent viral population growth only 20 years ago and is evolving at a much higher evolutionary rate than other genotypes infecting humans. This study constitutes mounting evidence of MastV-Sp6Gt7 active circulation and highlights the need for diagnostics capable of detecting it.
Keywords: Bayesian inference of phylogeny; Mamastrovirus; cospeciation; host-jump event; pairwise sequence comparison; zoonotic emergence.
Conflict of interest statement
LJP, KF, MGB, and GAC are all employees and Abbott shareholders.
Figures

Similar articles
-
Co-circulation of classic and novel astrovirus strains in patients with acute gastroenteritis in Germany.J Infect. 2018 May;76(5):457-464. doi: 10.1016/j.jinf.2018.02.006. Epub 2018 Feb 14. J Infect. 2018. PMID: 29454018
-
Diversity of Astrovirus in Goats in Southwest China and Identification of Two Novel Caprine Astroviruses.Microbiol Spectr. 2022 Aug 31;10(4):e0121822. doi: 10.1128/spectrum.01218-22. Epub 2022 Jul 7. Microbiol Spectr. 2022. PMID: 35862967 Free PMC article.
-
Diversity of human astroviruses in Germany 2018 and 2019.Virol J. 2022 Dec 21;19(1):221. doi: 10.1186/s12985-022-01955-3. Virol J. 2022. PMID: 36544187 Free PMC article.
-
Human astroviruses.Clin Microbiol Rev. 2014 Oct;27(4):1048-74. doi: 10.1128/CMR.00013-14. Clin Microbiol Rev. 2014. PMID: 25278582 Free PMC article. Review.
-
The changing epidemiology of astrovirus-associated gastroenteritis: a review.Arch Virol Suppl. 1996;12:287-300. doi: 10.1007/978-3-7091-6553-9_31. Arch Virol Suppl. 1996. PMID: 9015126 Review.
Cited by
-
Leveraging machine learning for taxonomic classification of emerging astroviruses.Front Mol Biosci. 2024 Jan 11;10:1305506. doi: 10.3389/fmolb.2023.1305506. eCollection 2023. Front Mol Biosci. 2024. PMID: 38274100 Free PMC article.
-
Mouse and human immune responses share neutralization epitopes of HAstV-VA1.J Virol. 2024 Jul 23;98(7):e0097124. doi: 10.1128/jvi.00971-24. Epub 2024 Jun 25. J Virol. 2024. PMID: 38916399 Free PMC article.
-
Ecological Changes Exacerbating the Spread of Invasive Ticks has Driven the Dispersal of Severe Fever with Thrombocytopenia Syndrome Virus Throughout Southeast Asia.Mol Biol Evol. 2024 Aug 2;41(8):msae173. doi: 10.1093/molbev/msae173. Mol Biol Evol. 2024. PMID: 39191515 Free PMC article.
-
Emergence, persistence, and positive selection of yellow fever virus in Colombia.Front Microbiol. 2025 Apr 7;16:1548556. doi: 10.3389/fmicb.2025.1548556. eCollection 2025. Front Microbiol. 2025. PMID: 40260085 Free PMC article.
-
A novel post-mortem pathogen discovery program detects an outbreak of Echovirus E7: Uganda, 2022-2023.Front Microbiol. 2025 Jul 1;16:1557576. doi: 10.3389/fmicb.2025.1557576. eCollection 2025. Front Microbiol. 2025. PMID: 40666800 Free PMC article.
References
-
- Appleton H, Higgins PG.. Letter: viruses and gastroenteritis in infants. Lancet (London, England). 1975 Jun 7;1(7919):1297. - PubMed
-
- Chapter 27 - Caliciviridae and Astroviridae. In: MacLachlan NJ, Dubovi EJ, editors. Fenner's veterinary virology. 5th ed. Boston: Academic Press; 2017. p. 497–510.
-
- Family - Astroviridae. In: King AMQ, Adams MJ, Carstens EB, et al., editors. Virus taxonomy. San Diego: Elsevier; 2012. p. 953–959.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical