

Mechanisms of NLRP3 inflammasome-mediated hepatic stellate cell activation: Therapeutic potential for liver fibrosis
- PMID: 37223529
- PMCID: PMC10201559
- DOI: 10.1016/j.gendis.2021.12.006
Mechanisms of NLRP3 inflammasome-mediated hepatic stellate cell activation: Therapeutic potential for liver fibrosis
Abstract
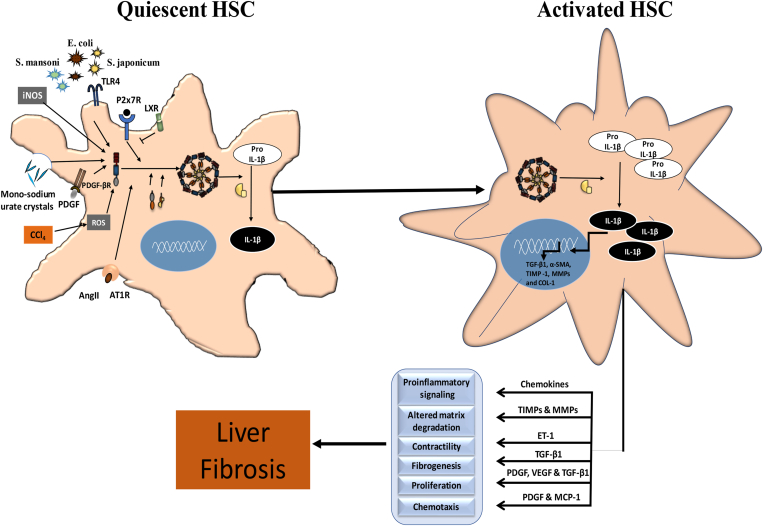
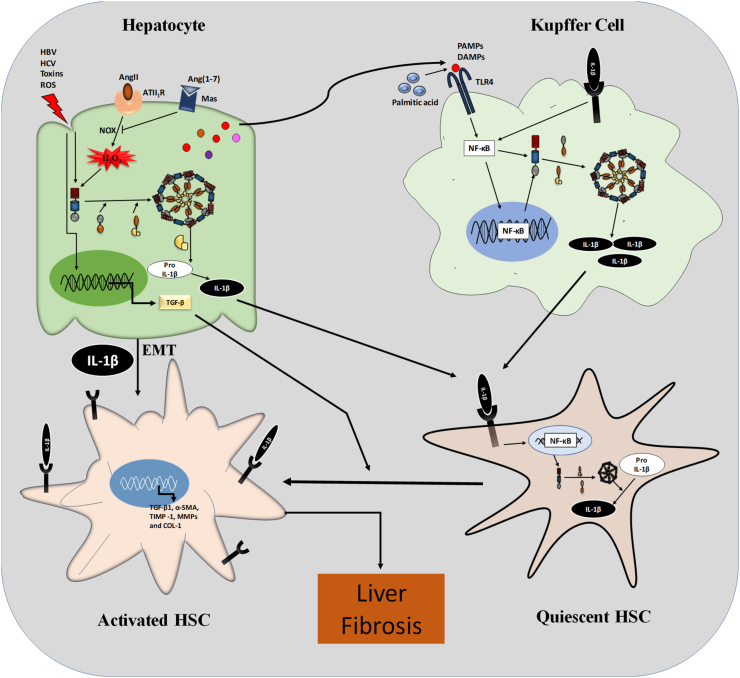
The liver injury leads to an inflammatory response, which causes the activation of hepatic stellate cells (HSCs) that further secrete ECM proteins and play an important role in liver fibrosis. Moreover, the inflammatory response is a driving force for fibrogenesis, which is triggered by many types of injuries. Exaggerated inflammatory immune responses are mediated by cytoplasmic protein complexes known as inflammasomes, which are involved in many chronic liver diseases. Inflammasomes are pattern recognition receptors (PRRs) that can sense any microbial motifs known as pathogen-associated molecular patterns (PAMPs), and host- or environmental-derived stress signals known as damage-associated molecular patterns (DAMPs). The inflammasomes cause caspase-mediated proteolytic cleavage of pro-IL-1β and pro-IL-18 into active IL-1β and IL-18. In this review, we provide a comprehensive summary of the important roles of NLRP3 inflammasome in the pathogenesis of liver fibrosis with an emphasis on several direct and indirect pathways responsible for the NLRP3 inflammasome-mediated HSCs activation and fibrogenesis. In addition, we discuss the general pharmacological and genetics strategies for the inhibition of NLRP3 inflammasome activation and its downstream signaling with examples of emerging pharmacotherapeutics, targeting the NLRP3 inflammasome signaling as well as a possible way to develop effective and safer NLRP3 inflammasome inhibitors.
Keywords: Hepatic stellate cells; Liver fibrosis; NLRP3 activation; NLRP3 inflammasome; NLRP3 inhibitors.
© 2022 The Authors. Publishing services by Elsevier B.V. on behalf of KeAi Communications Co., Ltd.
Figures

References
-
- Friedman S.L. Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1(2):98–105. - PubMed
-
- World Health Organization . 2018. The top 10 causes of death: leading causes of death by economy income group.http://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-...
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
