

Unraveling the small proteome of the plant symbiont Sinorhizobium meliloti by ribosome profiling and proteogenomics
- PMID: 37223733
- PMCID: PMC10117765
- DOI: 10.1093/femsml/uqad012
Unraveling the small proteome of the plant symbiont Sinorhizobium meliloti by ribosome profiling and proteogenomics
Erratum in
-
Correction to: Unraveling the small proteome of the plant symbiont Sinorhizobium meliloti by ribosome profiling and proteogenomics.Microlife. 2025 Mar 28;6:uqaf004. doi: 10.1093/femsml/uqaf004. eCollection 2025. Microlife. 2025. PMID: 40162304 Free PMC article.
Abstract
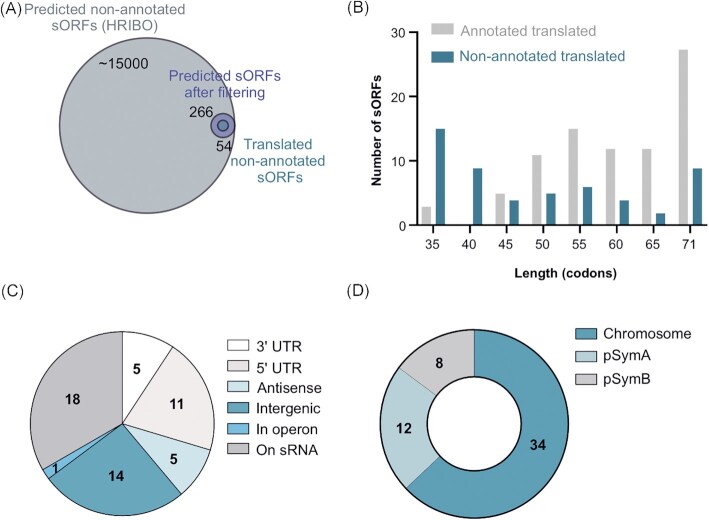
The soil-dwelling plant symbiont Sinorhizobium meliloti is a major model organism of Alphaproteobacteria. Despite numerous detailed OMICS studies, information about small open reading frame (sORF)-encoded proteins (SEPs) is largely missing, because sORFs are poorly annotated and SEPs are hard to detect experimentally. However, given that SEPs can fulfill important functions, identification of translated sORFs is critical for analyzing their roles in bacterial physiology. Ribosome profiling (Ribo-seq) can detect translated sORFs with high sensitivity, but is not yet routinely applied to bacteria because it must be adapted for each species. Here, we established a Ribo-seq procedure for S. meliloti 2011 based on RNase I digestion and detected translation for 60% of the annotated coding sequences during growth in minimal medium. Using ORF prediction tools based on Ribo-seq data, subsequent filtering, and manual curation, the translation of 37 non-annotated sORFs with ≤ 70 amino acids was predicted with confidence. The Ribo-seq data were supplemented by mass spectrometry (MS) analyses from three sample preparation approaches and two integrated proteogenomic search database (iPtgxDB) types. Searches against standard and 20-fold smaller Ribo-seq data-informed custom iPtgxDBs confirmed 47 annotated SEPs and identified 11 additional novel SEPs. Epitope tagging and Western blot analysis confirmed the translation of 15 out of 20 SEPs selected from the translatome map. Overall, by combining MS and Ribo-seq approaches, the small proteome of S. meliloti was substantially expanded by 48 novel SEPs. Several of them are part of predicted operons and/or are conserved from Rhizobiaceae to Bacteria, suggesting important physiological functions.
Keywords: Alphaproteobacteria; Ribosome profiling; Sinorhizobium meliloti; proteogenomics; proteomics; small open reading frame; small proteins.
© The Author(s) 2023. Published by Oxford University Press on behalf of FEMS.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures







Comment in
-
Correction to: Unraveling the small proteome of the plant symbiont Sinorhizobium meliloti by ribosome profiling and proteogenomics.Microlife. 2025 Mar 28;6:uqaf004. doi: 10.1093/femsml/uqaf004. eCollection 2025. Microlife. 2025. PMID: 40162304 Free PMC article.
Similar articles
-
Revealing the small proteome of Haloferax volcanii by combining ribosome profiling and small-protein optimized mass spectrometry.Microlife. 2023 Jan 16;4:uqad001. doi: 10.1093/femsml/uqad001. eCollection 2023. Microlife. 2023. PMID: 37223747 Free PMC article.
-
RiboReport - benchmarking tools for ribosome profiling-based identification of open reading frames in bacteria.Brief Bioinform. 2022 Mar 10;23(2):bbab549. doi: 10.1093/bib/bbab549. Brief Bioinform. 2022. PMID: 35037022 Free PMC article.
-
An update on sORFs.org: a repository of small ORFs identified by ribosome profiling.Nucleic Acids Res. 2018 Jan 4;46(D1):D497-D502. doi: 10.1093/nar/gkx1130. Nucleic Acids Res. 2018. PMID: 29140531 Free PMC article.
-
Short open reading frames (sORFs) and microproteins: an update on their identification and validation measures.J Biomed Sci. 2022 Mar 17;29(1):19. doi: 10.1186/s12929-022-00802-5. J Biomed Sci. 2022. PMID: 35300685 Free PMC article. Review.
-
Small Open Reading Frame-Encoded Micro-Peptides: An Emerging Protein World.Int J Mol Sci. 2023 Jun 23;24(13):10562. doi: 10.3390/ijms241310562. Int J Mol Sci. 2023. PMID: 37445739 Free PMC article. Review.
Cited by
-
Ribosome Profiling Methods Adapted to the Study of RNA-Dependent Translation Regulation in Staphylococcus aureus.Methods Mol Biol. 2024;2741:73-100. doi: 10.1007/978-1-0716-3565-0_5. Methods Mol Biol. 2024. PMID: 38217649
-
Complementary Ribo-seq approaches map the translatome and provide a small protein census in the foodborne pathogen Campylobacter jejuni.Nat Commun. 2025 Mar 30;16(1):3078. doi: 10.1038/s41467-025-58329-w. Nat Commun. 2025. PMID: 40159498 Free PMC article.
-
Uncovering the small proteome of Methanosarcina mazei using Ribo-seq and peptidomics under different nitrogen conditions.Nat Commun. 2024 Oct 6;15(1):8659. doi: 10.1038/s41467-024-53008-8. Nat Commun. 2024. PMID: 39370430 Free PMC article.
-
Detection of Known and Novel Small Proteins in Pseudomonas stutzeri Using a Combination of Bottom-Up and Digest-Free Proteomics and Proteogenomics.Anal Chem. 2023 Aug 15;95(32):11892-11900. doi: 10.1021/acs.analchem.3c00676. Epub 2023 Aug 3. Anal Chem. 2023. PMID: 37535005 Free PMC article.
-
Early posttranscriptional response to tetracycline exposure in a gram-negative soil bacterium reveals unexpected attenuation mechanism of a DUF1127 gene.RNA Biol. 2025 Dec;22(1):1-16. doi: 10.1080/15476286.2025.2521887. Epub 2025 Jul 1. RNA Biol. 2025. PMID: 40534149 Free PMC article.
References
-
- Bartel J, Varadarajan AR, Sura T et al. Optimized proteomics workflow for the detection of small proteins. J Proteome Res. 2020;19:4004–18. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases