

Elucidation of Short Linear Motif-Based Interactions of the FERM Domains of Ezrin, Radixin, Moesin, and Merlin
- PMID: 37224425
- PMCID: PMC10249358
- DOI: 10.1021/acs.biochem.3c00096
Elucidation of Short Linear Motif-Based Interactions of the FERM Domains of Ezrin, Radixin, Moesin, and Merlin
Abstract
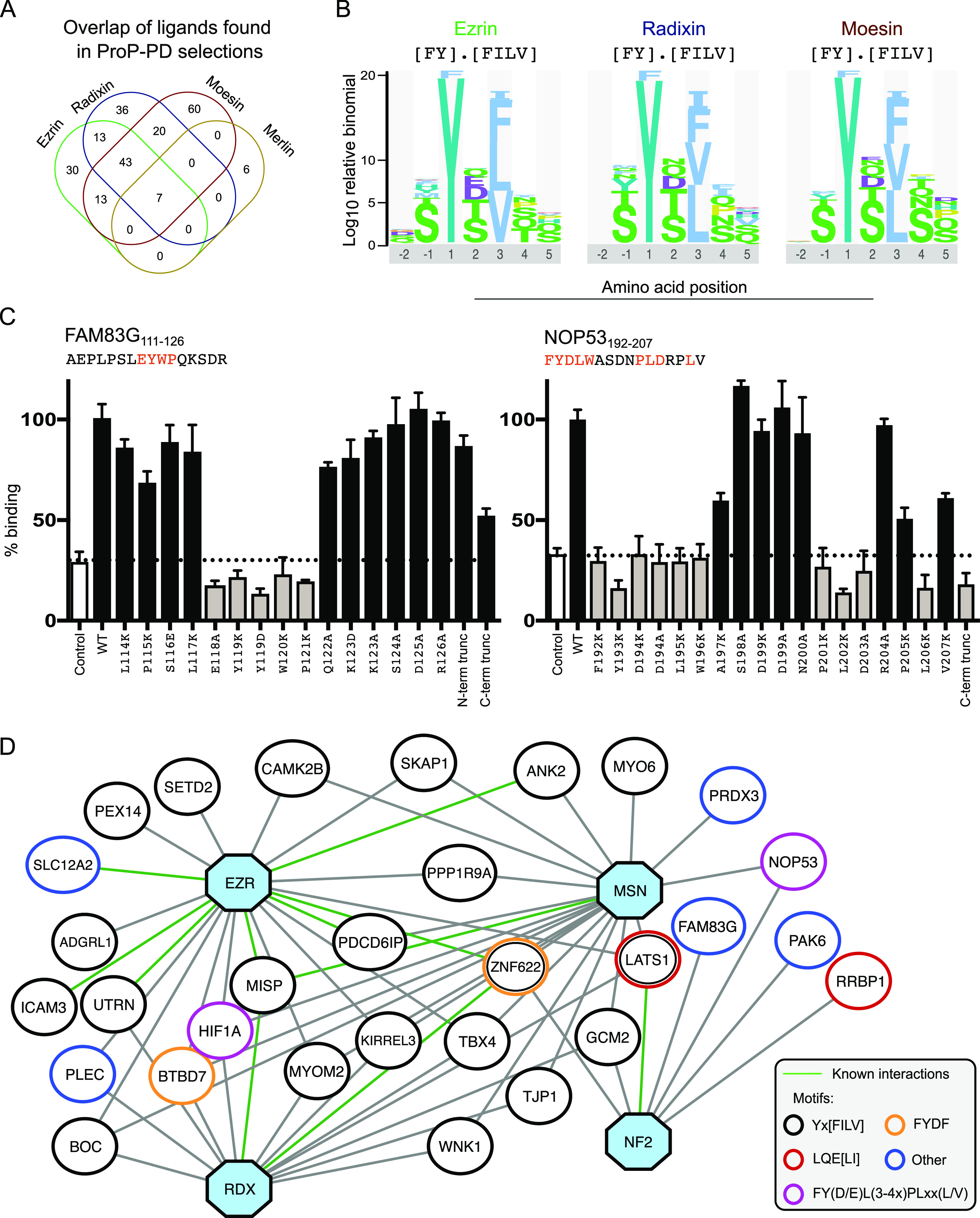
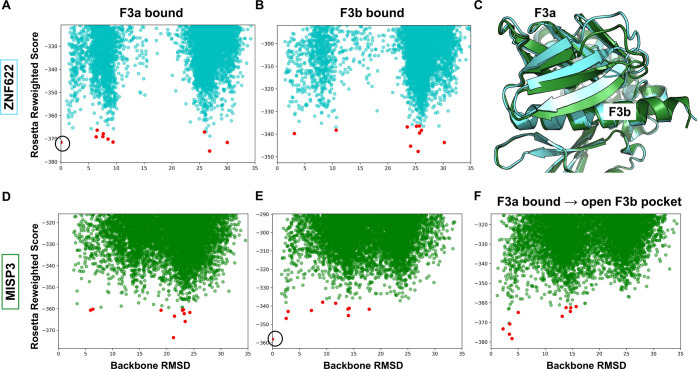
The ERM (ezrin, radixin, and moesin) family of proteins and the related protein merlin participate in scaffolding and signaling events at the cell cortex. The proteins share an N-terminal FERM [band four-point-one (4.1) ERM] domain composed of three subdomains (F1, F2, and F3) with binding sites for short linear peptide motifs. By screening the FERM domains of the ERMs and merlin against a phage library that displays peptides representing the intrinsically disordered regions of the human proteome, we identified a large number of novel ligands. We determined the affinities for the ERM and merlin FERM domains interacting with 18 peptides and validated interactions with full-length proteins through pull-down experiments. The majority of the peptides contained an apparent Yx[FILV] motif; others show alternative motifs. We defined distinct binding sites for two types of similar but distinct binding motifs (YxV and FYDF) using a combination of Rosetta FlexPepDock computational peptide docking protocols and mutational analysis. We provide a detailed molecular understanding of how the two types of peptides with distinct motifs bind to different sites on the moesin FERM phosphotyrosine binding-like subdomain and uncover interdependencies between the different types of ligands. The study expands the motif-based interactomes of the ERMs and merlin and suggests that the FERM domain acts as a switchable interaction hub.
Conflict of interest statement
The authors declare no competing financial interest.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
