

Deep phenotypic characterization of the retinal dystrophy in patients with RNU4ATAC-associated Roifman syndrome
- PMID: 37225827
- PMCID: PMC10697969
- DOI: 10.1038/s41433-023-02581-1
Deep phenotypic characterization of the retinal dystrophy in patients with RNU4ATAC-associated Roifman syndrome
Abstract
Purpose: To characterize the retinal phenotype in RNU4ATAC-associated Roifman syndrome.
Methods: Ten patients (including 8 males) with molecularly confirmed Roifman syndrome underwent detailed ophthalmologic evaluation including fundus imaging, fundus autofluorescence (FAF) imaging, spectral-domain optical coherence tomography (SD-OCT), and electroretinography (ERG). Six patients had follow-up eye exams. All patients also underwent comprehensive examination for features of extra-retinal Roifman syndrome.
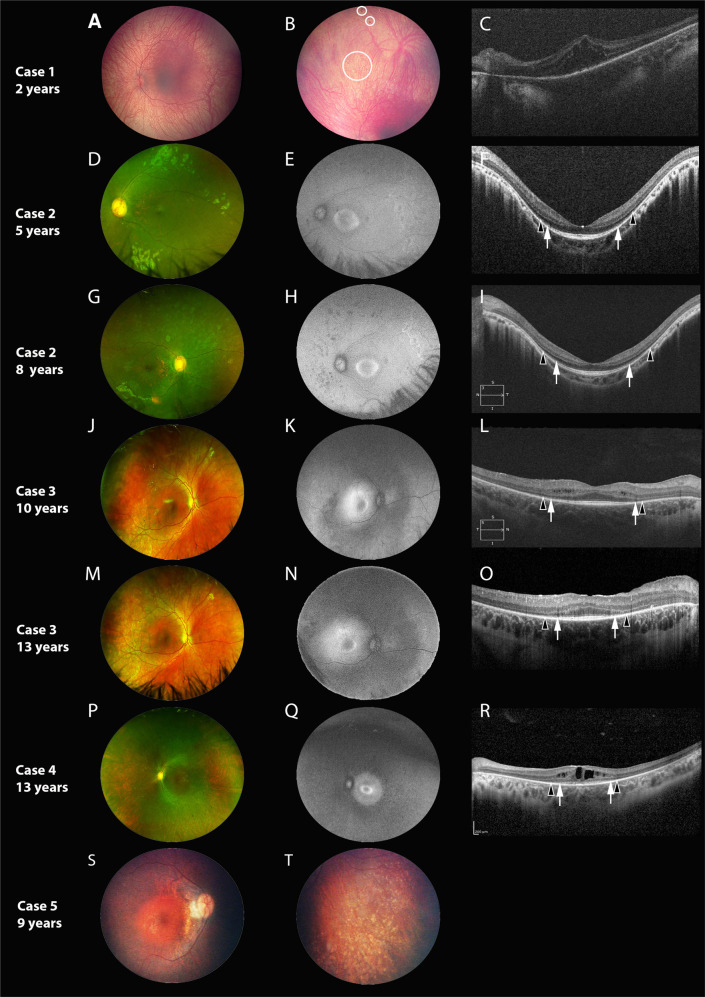
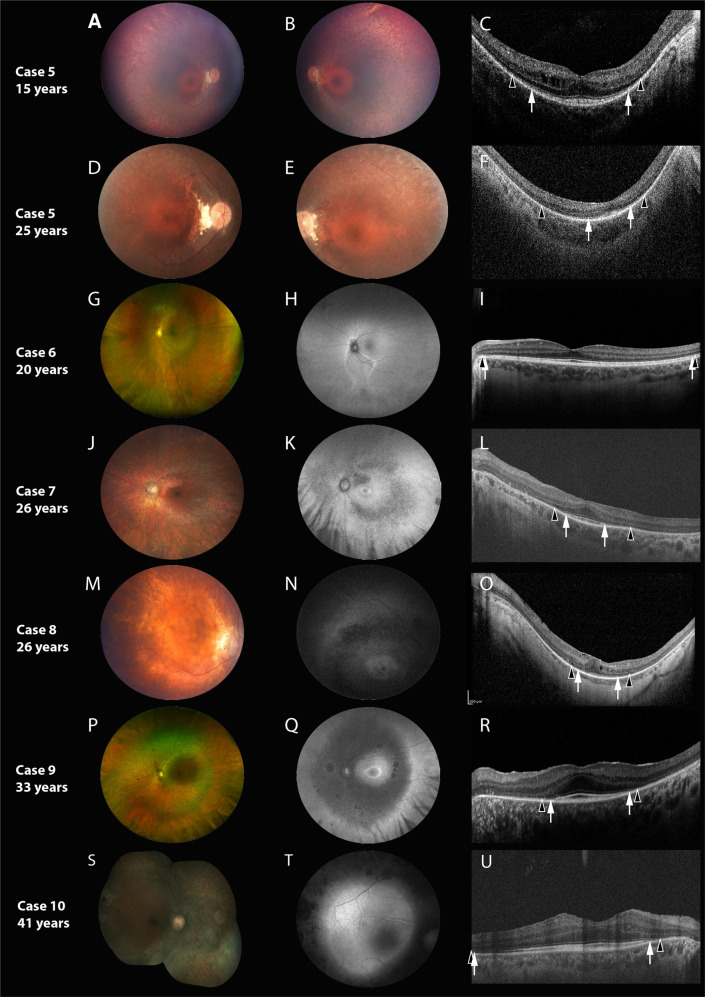
Results: All patients had biallelic RNU4ATAC variants. Nyctalopia was common (7/10). Visual acuity at presentation ranged from 20/20 to 20/200 (Age Range: 5-41 years). Retinal exam revealed features of generalized retinopathy with mid-peripheral pigment epithelial changes. A para or peri-foveal ring of hyper-autofluorescence was the commonest FAF abnormality noted (6/8). The SD-OCT demonstrated relative preservation of the foveal ellipsoid zone in six cases; associated features included cystoid changes (5/10) and posterior staphyloma (3/10). The ERG was abnormal in all patients; nine showed generalized rod-cone dystrophy, whilst one patient with sectoral retinal involvement only had isolated rod dystrophy (20 years old). On follow-up examination (Mean duration: 8.16 years), progressive loss of visual acuity (2/6), mid-peripheral retinal atrophy (3/6) or shortening of ellipsoid zone width (1/6) were observed.
Conclusion: This study has characterized the retinal phenotype in RNU4ATAC-associated Roifman syndrome. Retinal involvement is universal, early-onset, and overall, the retinal and FAF features are consistent with rod-cone degeneration that is slowly progressive over time. The sub-foveal retinal ultrastructure is relatively preserved in majority of patients. Phenotypic variability independent of age exists, and more study of allelic- and sex-based determinants of disease severity are necessary.
© 2023. Crown.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Abdel-Salam GM, Miyake N, Eid MM, Abdel-Hamid MS, Hassan NA, Eid OM, et al. A homozygous mutation in RNU4ATAC as a cause of microcephalic osteodysplastic primordial dwarfism type I (MOPD I) with associated pigmentary disorder. Am J Med Genet A. 2011;155A:2885–96. doi: 10.1002/ajmg.a.34299. - DOI - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
