

A small-molecule PI3Kα activator for cardioprotection and neuroregeneration
- PMID: 37225977
- PMCID: PMC7614683
- DOI: 10.1038/s41586-023-05972-2
A small-molecule PI3Kα activator for cardioprotection and neuroregeneration
Abstract
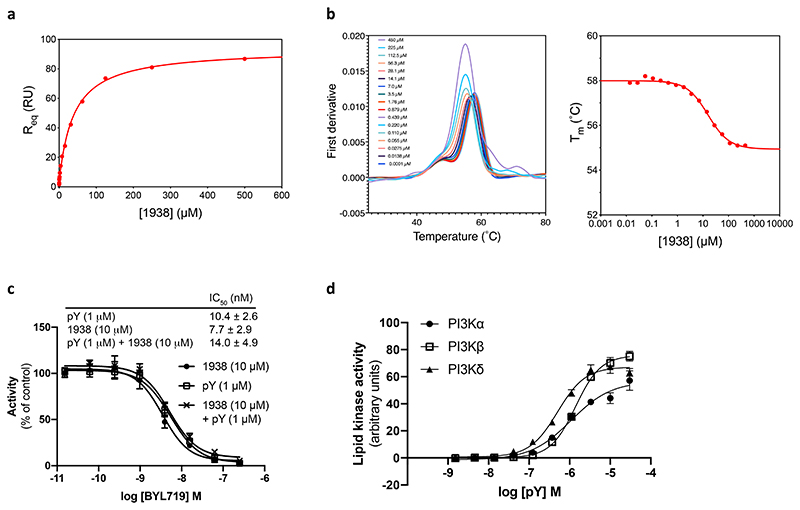
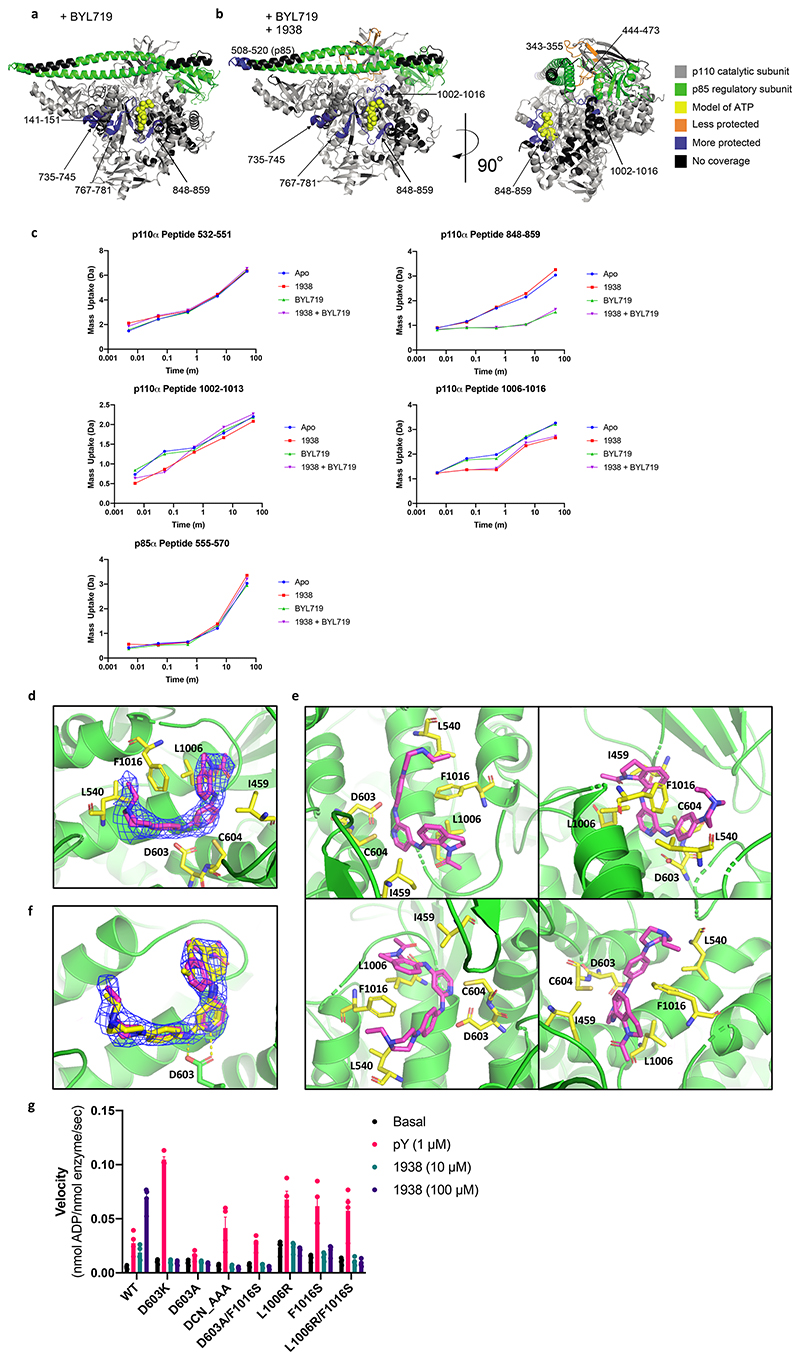
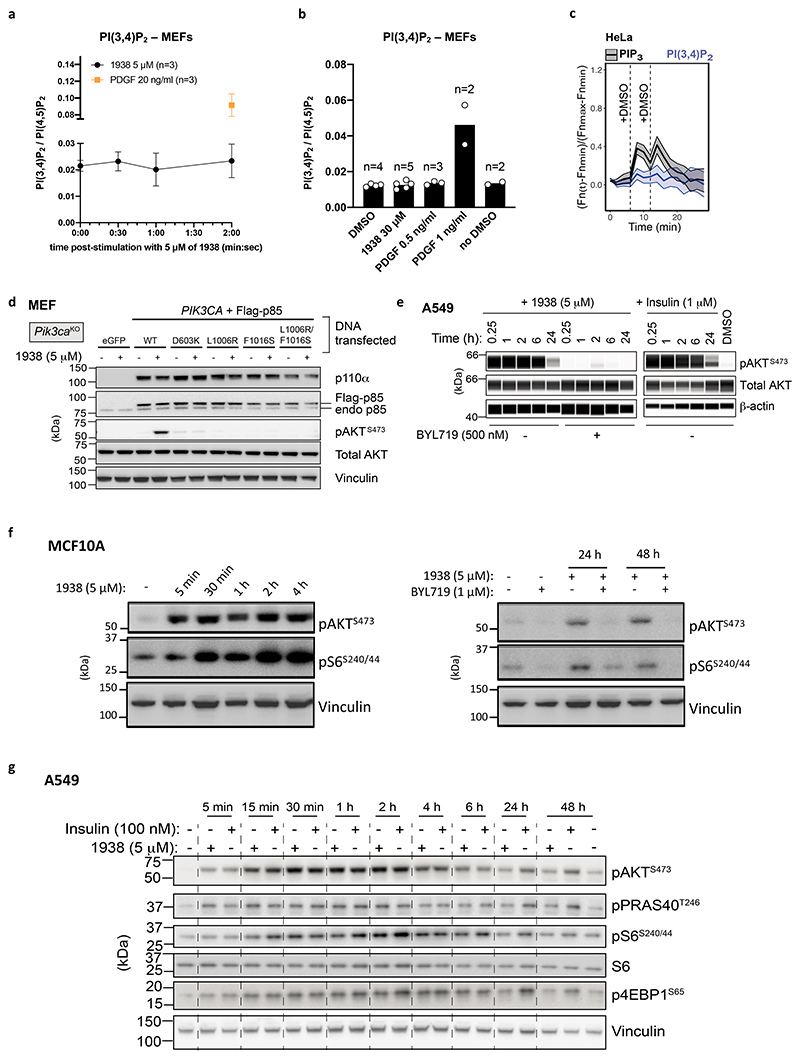
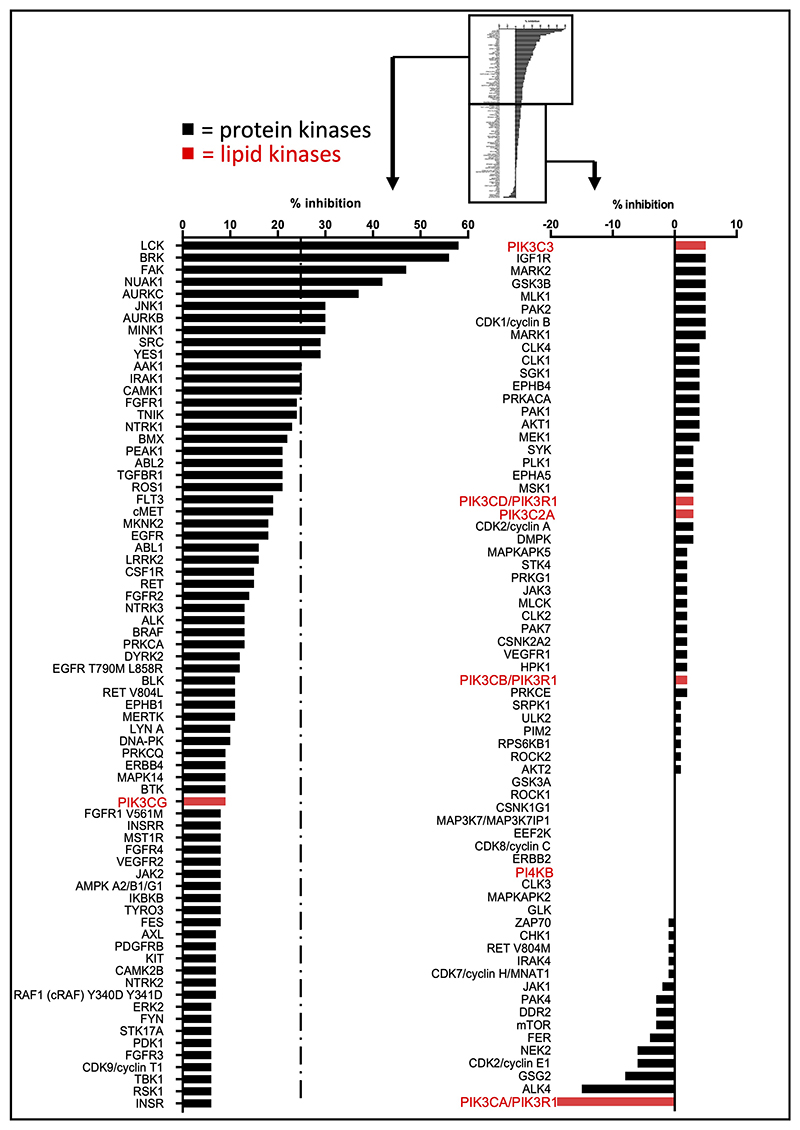
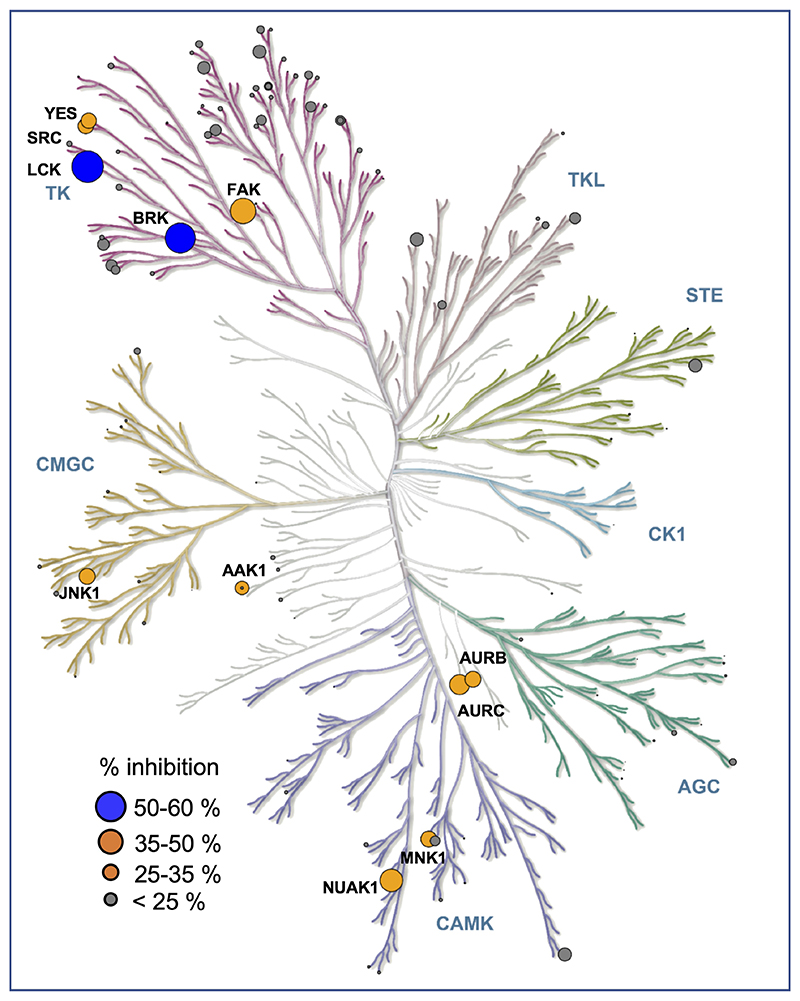
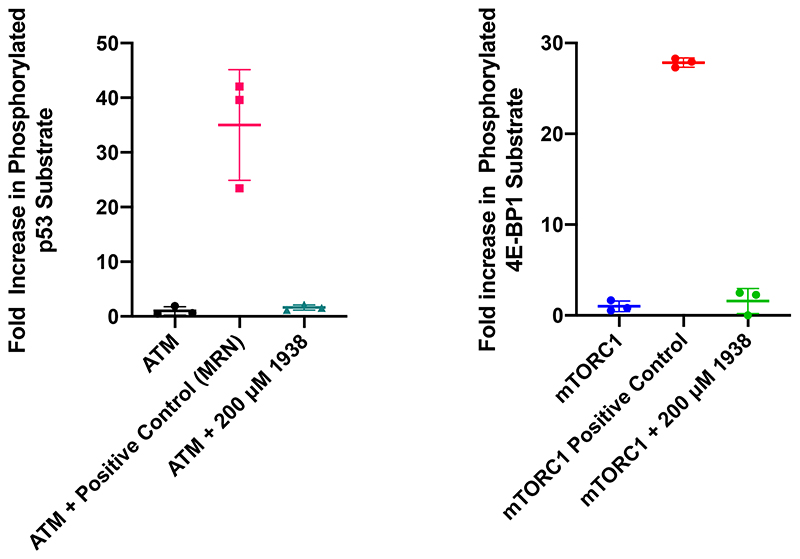
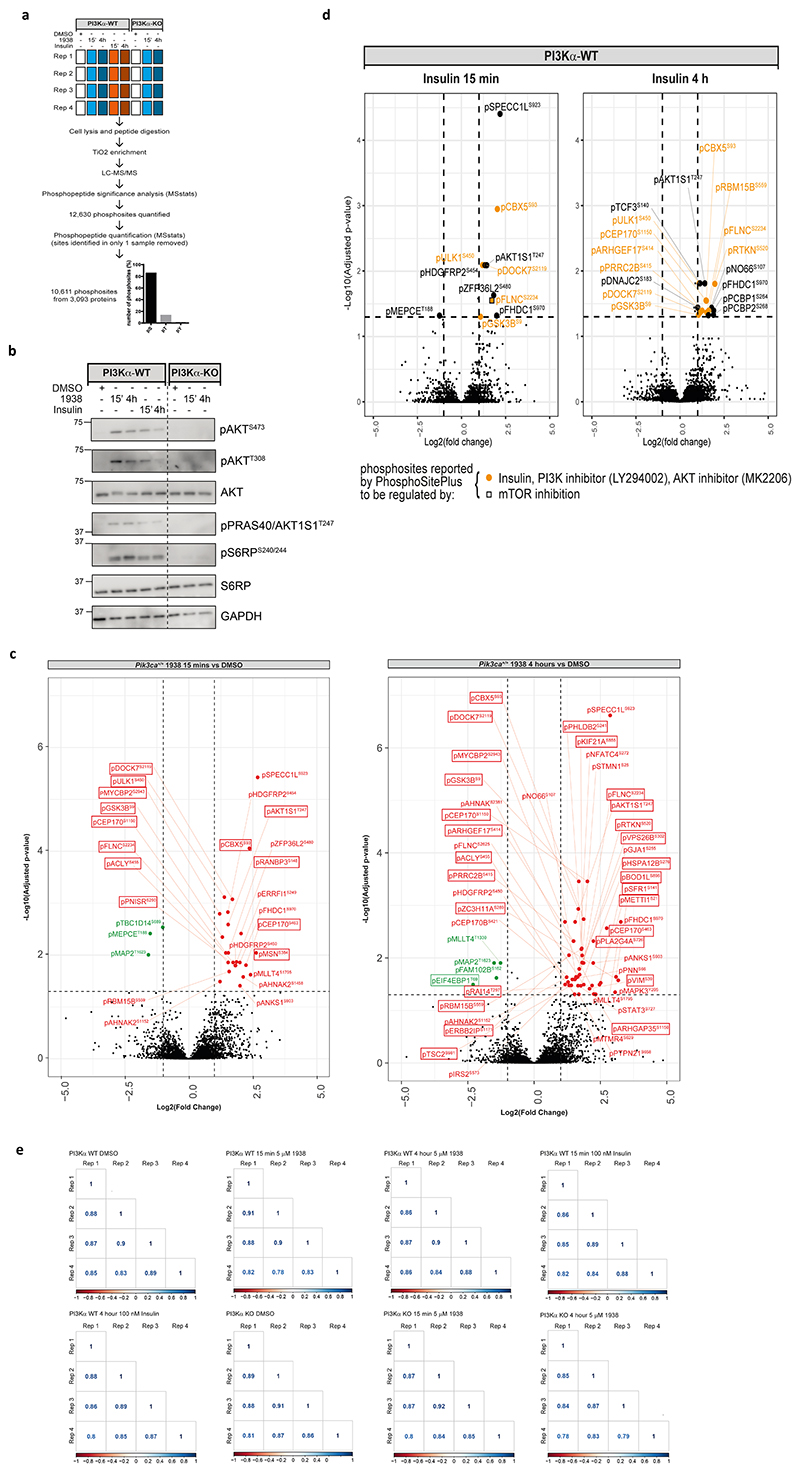
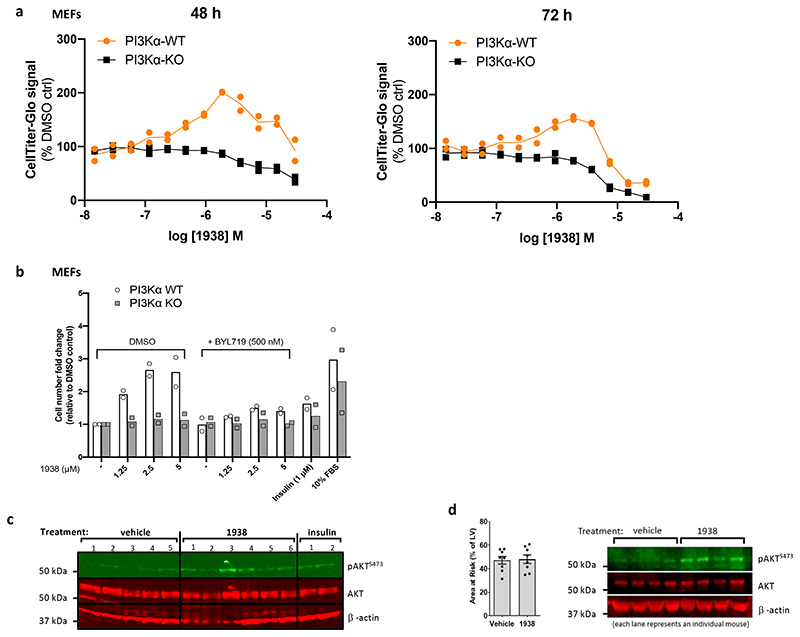
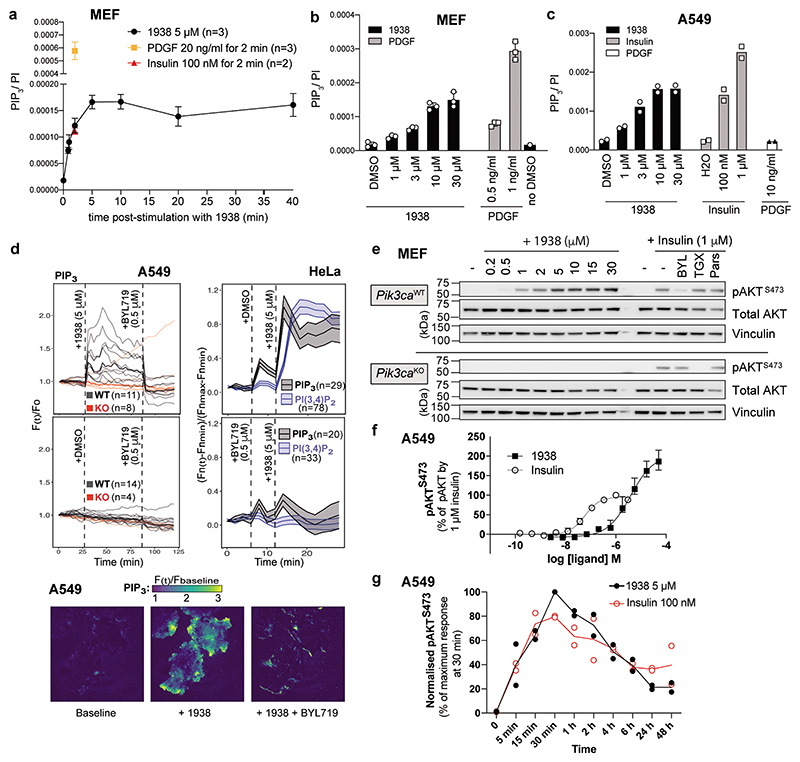
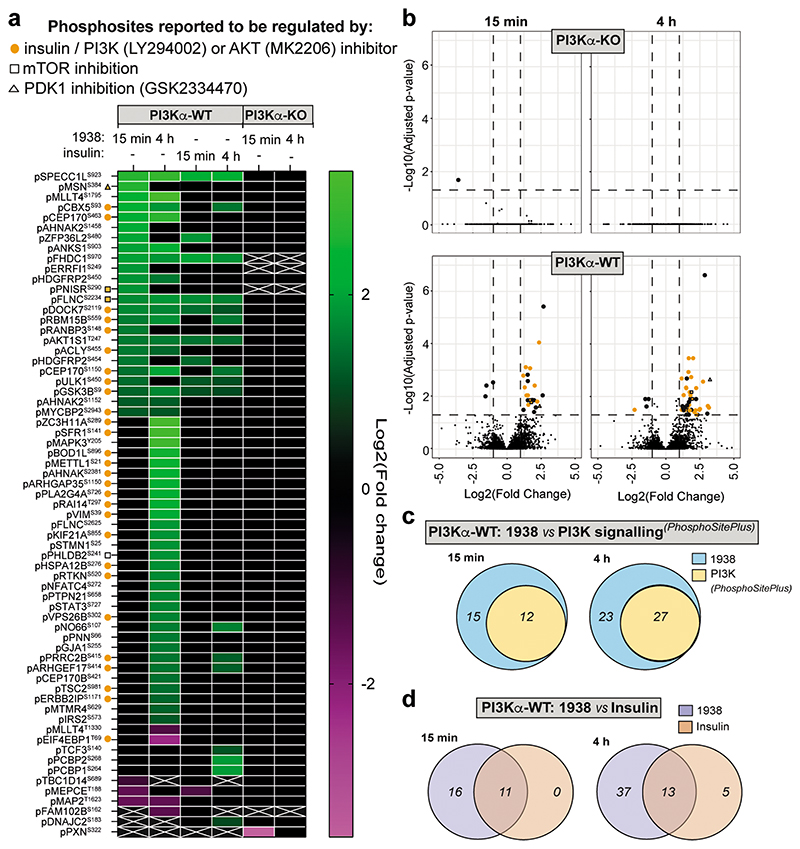
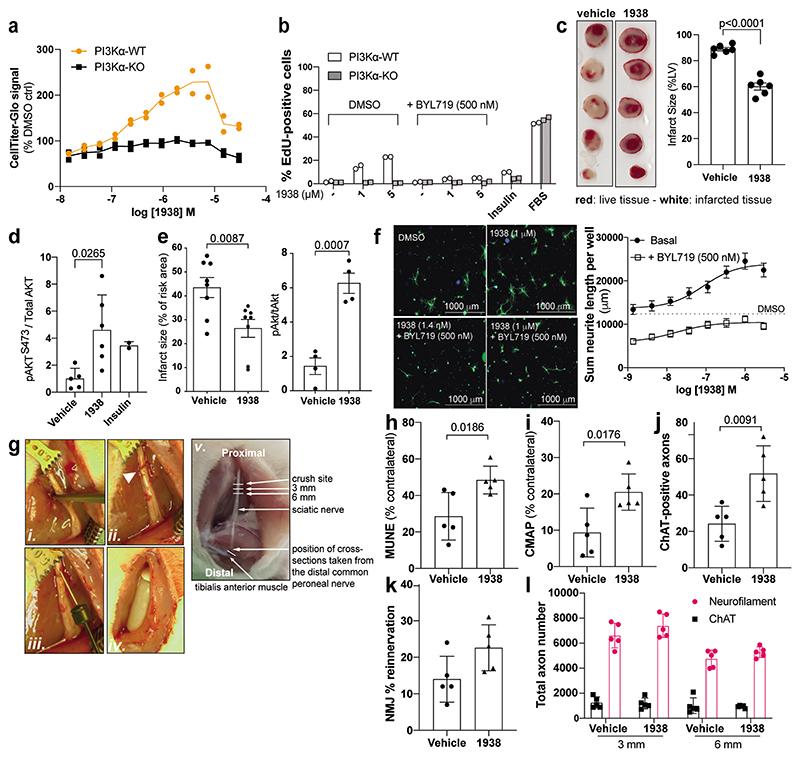
Harnessing the potential beneficial effects of kinase signalling through the generation of direct kinase activators remains an underexplored area of drug development1-5. This also applies to the PI3K signalling pathway, which has been extensively targeted by inhibitors for conditions with PI3K overactivation, such as cancer and immune dysregulation. Here we report the discovery of UCL-TRO-1938 (referred to as 1938 hereon), a small-molecule activator of the PI3Kα isoform, a crucial effector of growth factor signalling. 1938 allosterically activates PI3Kα through a distinct mechanism by enhancing multiple steps of the PI3Kα catalytic cycle and causes both local and global conformational changes in the PI3Kα structure. This compound is selective for PI3Kα over other PI3K isoforms and multiple protein and lipid kinases. It transiently activates PI3K signalling in all rodent and human cells tested, resulting in cellular responses such as proliferation and neurite outgrowth. In rodent models, acute treatment with 1938 provides cardioprotection from ischaemia-reperfusion injury and, after local administration, enhances nerve regeneration following nerve crush. This study identifies a chemical tool to directly probe the PI3Kα signalling pathway and a new approach to modulate PI3K activity, widening the therapeutic potential of targeting these enzymes through short-term activation for tissue protection and regeneration. Our findings illustrate the potential of activating kinases for therapeutic benefit, a currently largely untapped area of drug development.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
B.V. is a consultant for iOnctura (Geneva, Switzerland), Venthera (Palo Alto, US), Pharming (Leiden, the Netherlands) and Olema Pharmaceuticals (San Francisco, US), and has received speaker fees from Gilead (Foster City, US). M.F., M.C., I.F., A.M. and D.M.S. are or were employees and shareholders in AstraZeneca at the time of the work done. J.B.P. is co-Founder and Chief Scientific Officer of the UCL spinout company Glialign Ltd. The other authors do not have competing interests to disclose. A patent application GB 2113079.4, with relevance to this work has been filed by UCL Business and we want to declare our relationship with this patent application.
Figures
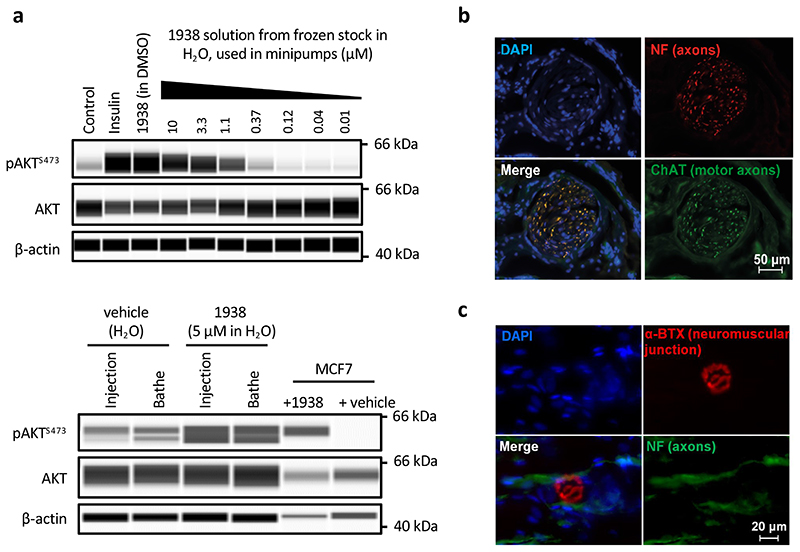
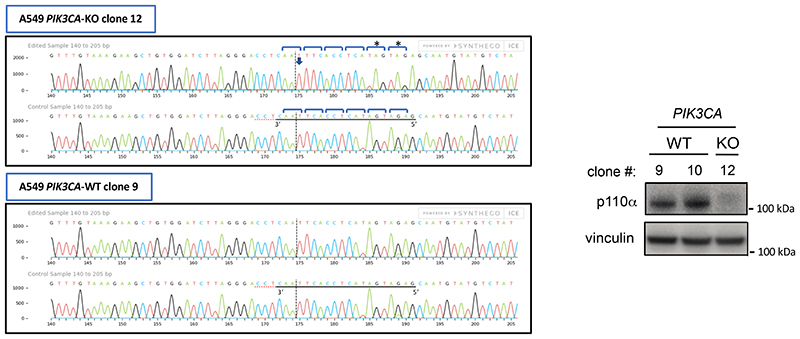
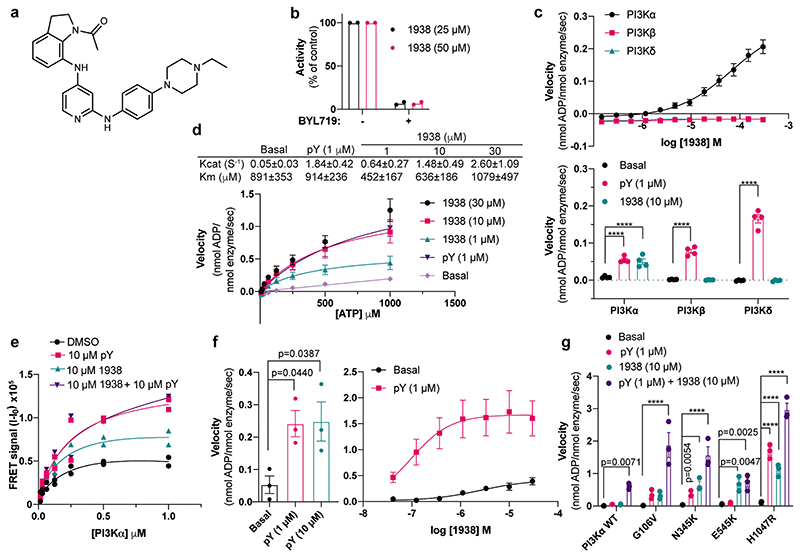
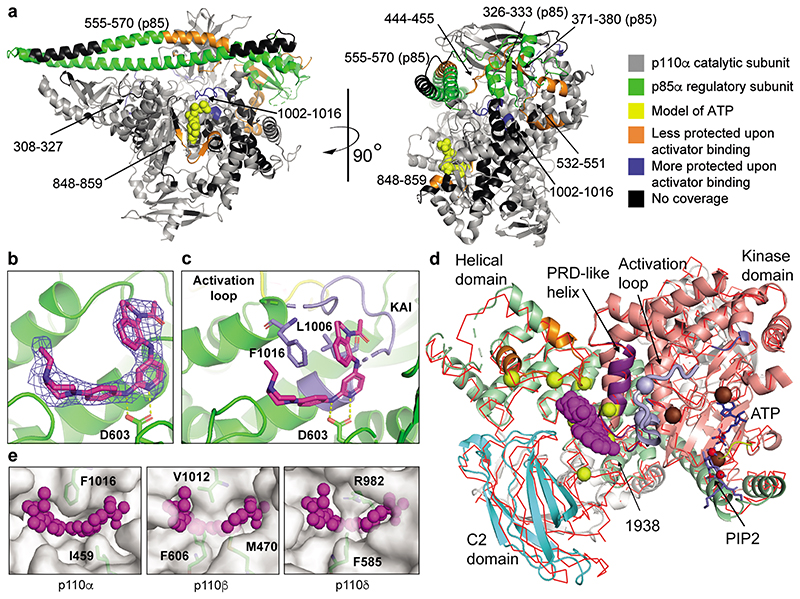
Comment in
-
PI3K activator taps route to regeneration.Nat Rev Drug Discov. 2023 Jul;22(7):537. doi: 10.1038/d41573-023-00097-7. Nat Rev Drug Discov. 2023. PMID: 37291416 No abstract available.
References
-
- Simpson GL, Hughes JA, Washio Y, Bertrand SM. Direct small-molecule kinase activation: Novel approaches for a new era of drug discovery. Curr Opin Drug Discov Devel. 2009;12:585–596. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- MC_PC_16063/MRC_/Medical Research Council/United Kingdom
- MC_PC_17202/MRC_/Medical Research Council/United Kingdom
- MC_UP_1201/6/MRC_/Medical Research Council/United Kingdom
- 25722/CRUK_/Cancer Research UK/United Kingdom
- MC_PC_18063/MRC_/Medical Research Council/United Kingdom
- BBS/E/B/000C0415/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- A20265/CRUK_/Cancer Research UK/United Kingdom
- MC_PC_15063/MRC_/Medical Research Council/United Kingdom
- 220464/WT_/Wellcome Trust/United Kingdom
- MC_U105184308/MRC_/Medical Research Council/United Kingdom
- MC_PC_16087/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
