

Genomic epidemiology of Staphylococcus aureus isolated from bloodstream infections in South America during 2019 supports regional surveillance
- PMID: 37227244
- PMCID: PMC10272885
- DOI: 10.1099/mgen.0.001020
Genomic epidemiology of Staphylococcus aureus isolated from bloodstream infections in South America during 2019 supports regional surveillance
Abstract
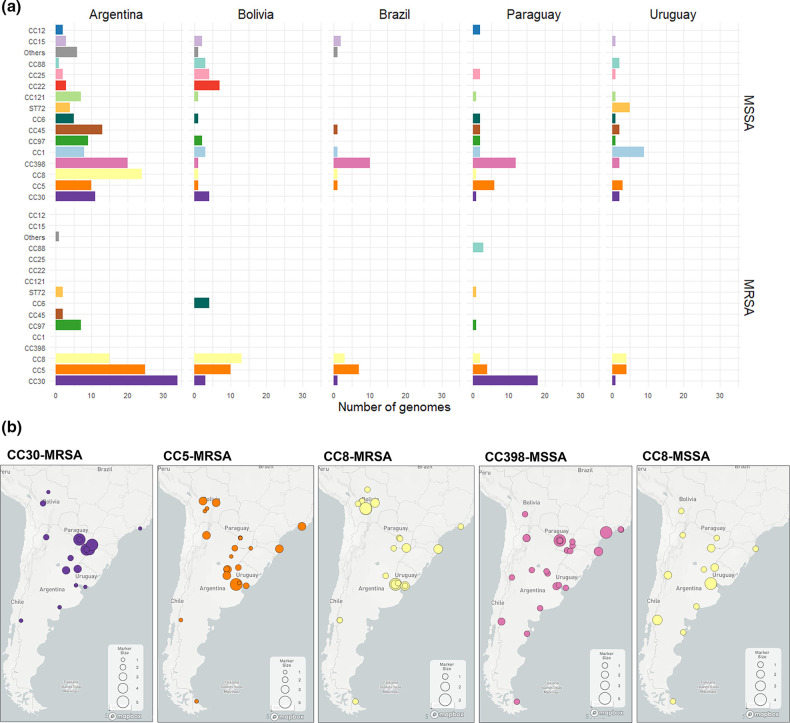
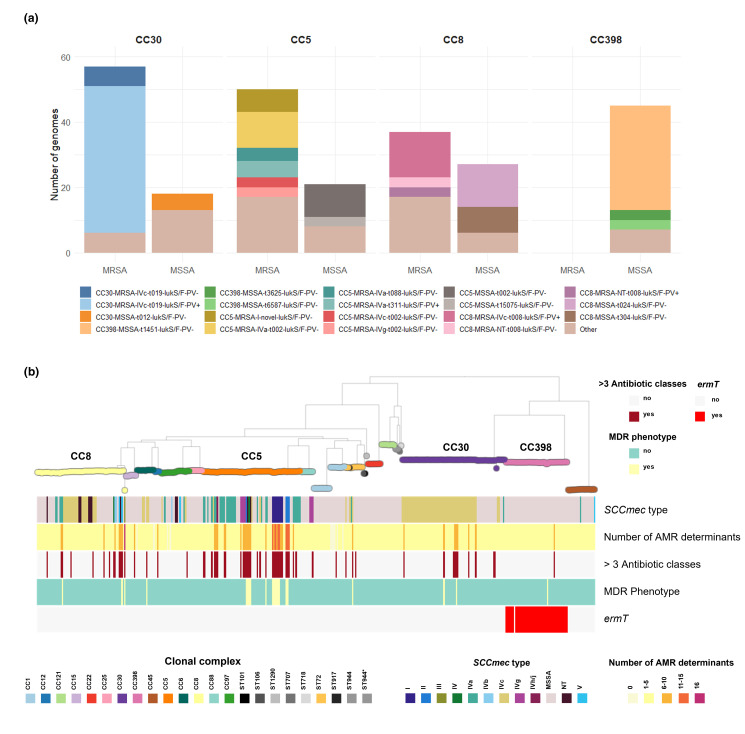
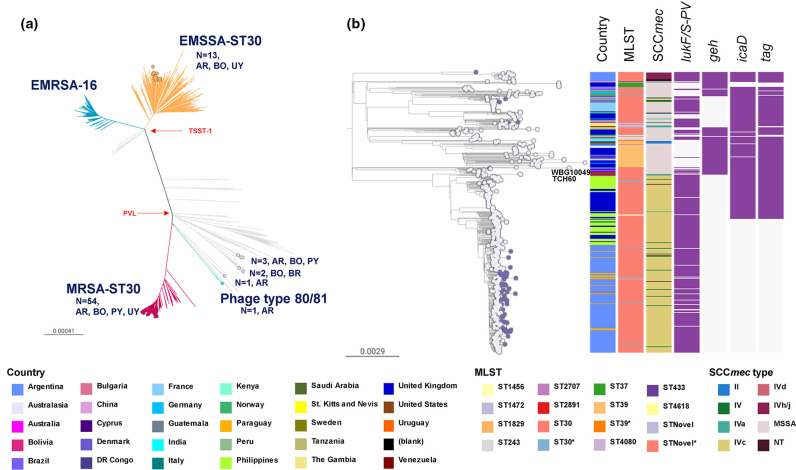
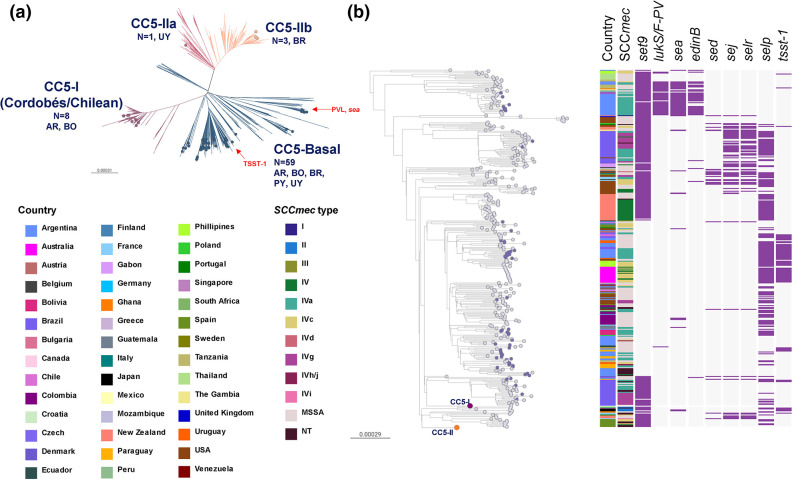
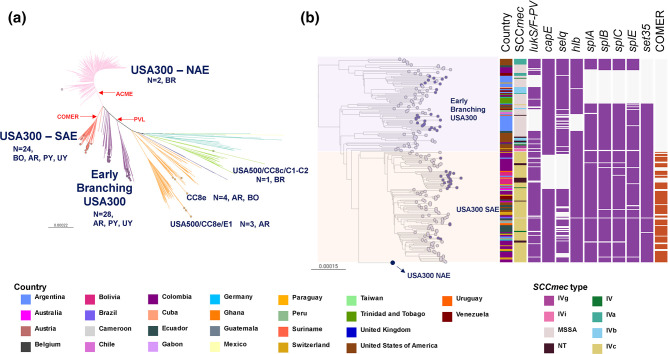
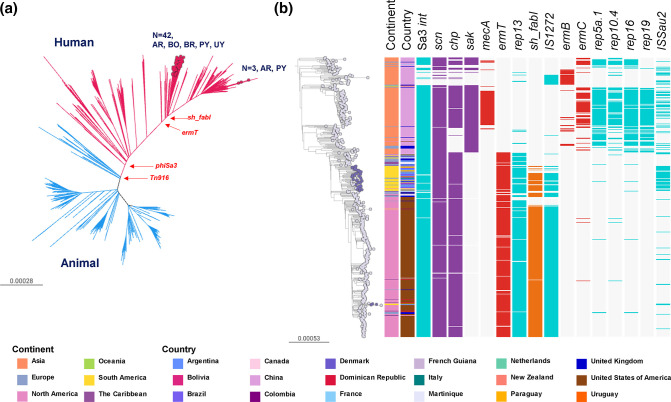
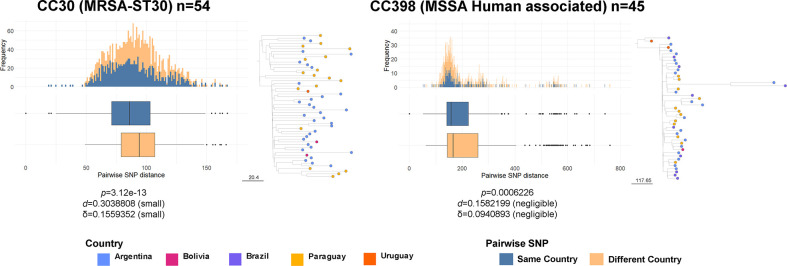
Staphylococcus aureus remains one of the leading causes of infections worldwide and a common cause of bacteraemia. However, studies documenting the epidemiology of S. aureus in South America using genomics are scarce. We hereby report on the largest genomic epidemiology study to date of both methicillin-resistant S. aureus (MRSA) and methicillin-susceptible S. aureus (MSSA) in South America, conducted by the StaphNET-SA network. We characterised 404 genomes recovered from a prospective observational study of S. aureus bacteraemia in 58 hospitals from Argentina, Bolivia, Brazil, Paraguay and Uruguay between April and October 2019. We show that a minority of S. aureus isolates are phenotypically multi-drug resistant (5.2%), but more than a quarter are resistant to macrolide-lincosamide-streptogramin B (MLSb). MSSA were more genetically diverse than MRSA. Lower rates of associated antimicrobial resistance in community-associated(CA)-MRSA versus hospital-associated (HA)-MRSA were found in association with three S. aureus genotypes dominating the MRSA population: CC30-MRSA-IVc-t019-lukS/F-PV+, CC5-MRSA-IV-t002-lukS/F-PV- and CC8-MRSA-IVc-t008-lukS/F-PV+-COMER+. These are historically from a CA origin, carry on average fewer antimicrobial resistance determinants, and often lack key virulence genes. Surprisingly, CC398-MSSA-t1451-lukS/F-PV- related to the CC398 human-associated lineage is widely disseminated throughout the region, and is described here for the first time as the most prevalent MSSA lineage in South America. Moreover, CC398 strains carrying ermT (largely responsible for the MLSb resistance rates of MSSA strains: inducible iMLSb phenotype) and sh_fabI (related to triclosan resistance) were recovered from both CA and HA origin. The frequency of MRSA and MSSA lineages differed between countries but the most prevalent S. aureus genotypes are high-risk clones widely distributed in the South American region without a clear country-specific phylogeographical structure. Therefore, our findings underline the need for continuous genomic surveillance by regional networks such as StaphNET-SA. This article contains data hosted by Microreact.
Keywords: CC30; CC398; CC5; CC8; MRSA; MSSA; S. aureus; South America.
Conflict of interest statement
We declare no conflicts of interest.
Figures
References
-
- WHO Antimicrobial resistance. Global report on surveillance. n.d.
-
- Bal AM, Coombs GW, Holden MTG, Lindsay JA, Nimmo GR, et al. Genomic insights into the emergence and spread of international clones of healthcare-, community- and livestock-associated meticillin-resistant Staphylococcus aureus: blurring of the traditional definitions. J Glob Antimicrob Resist. 2016;6:95–101. doi: 10.1016/j.jgar.2016.04.004. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous