

HDL Function and Atherosclerosis: Reactive Dicarbonyls as Promising Targets of Therapy
- PMID: 37228232
- PMCID: PMC10213997
- DOI: 10.1161/CIRCRESAHA.123.321563
HDL Function and Atherosclerosis: Reactive Dicarbonyls as Promising Targets of Therapy
Abstract
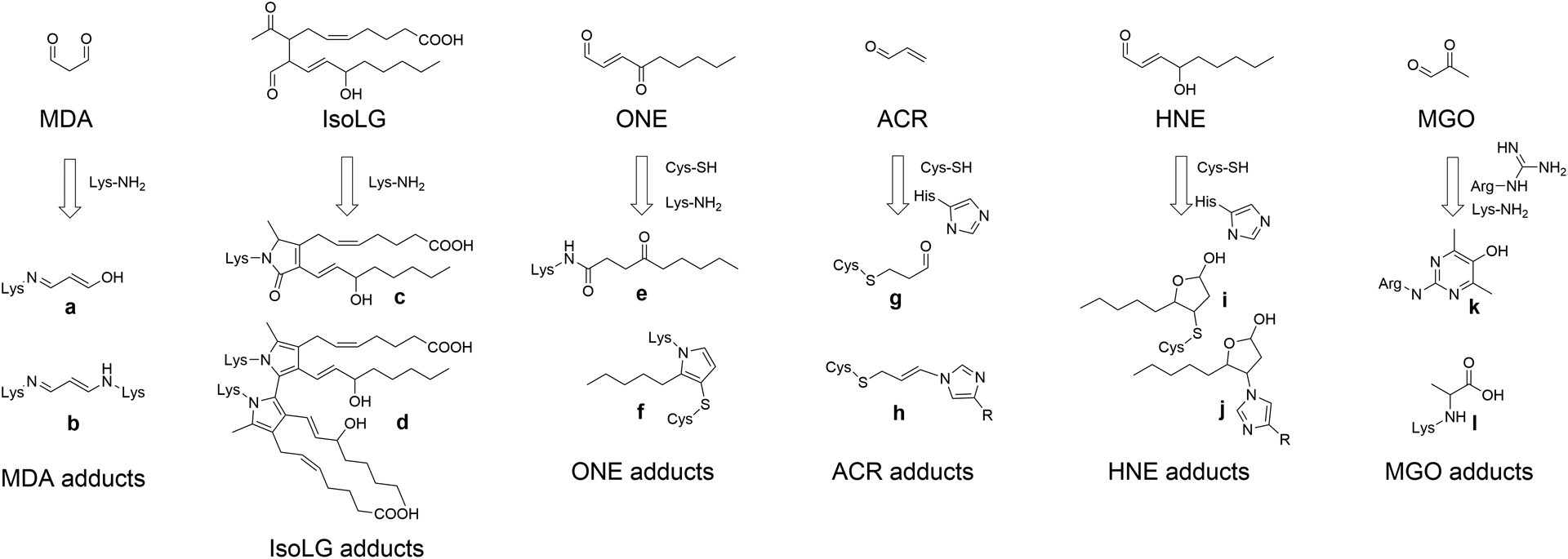
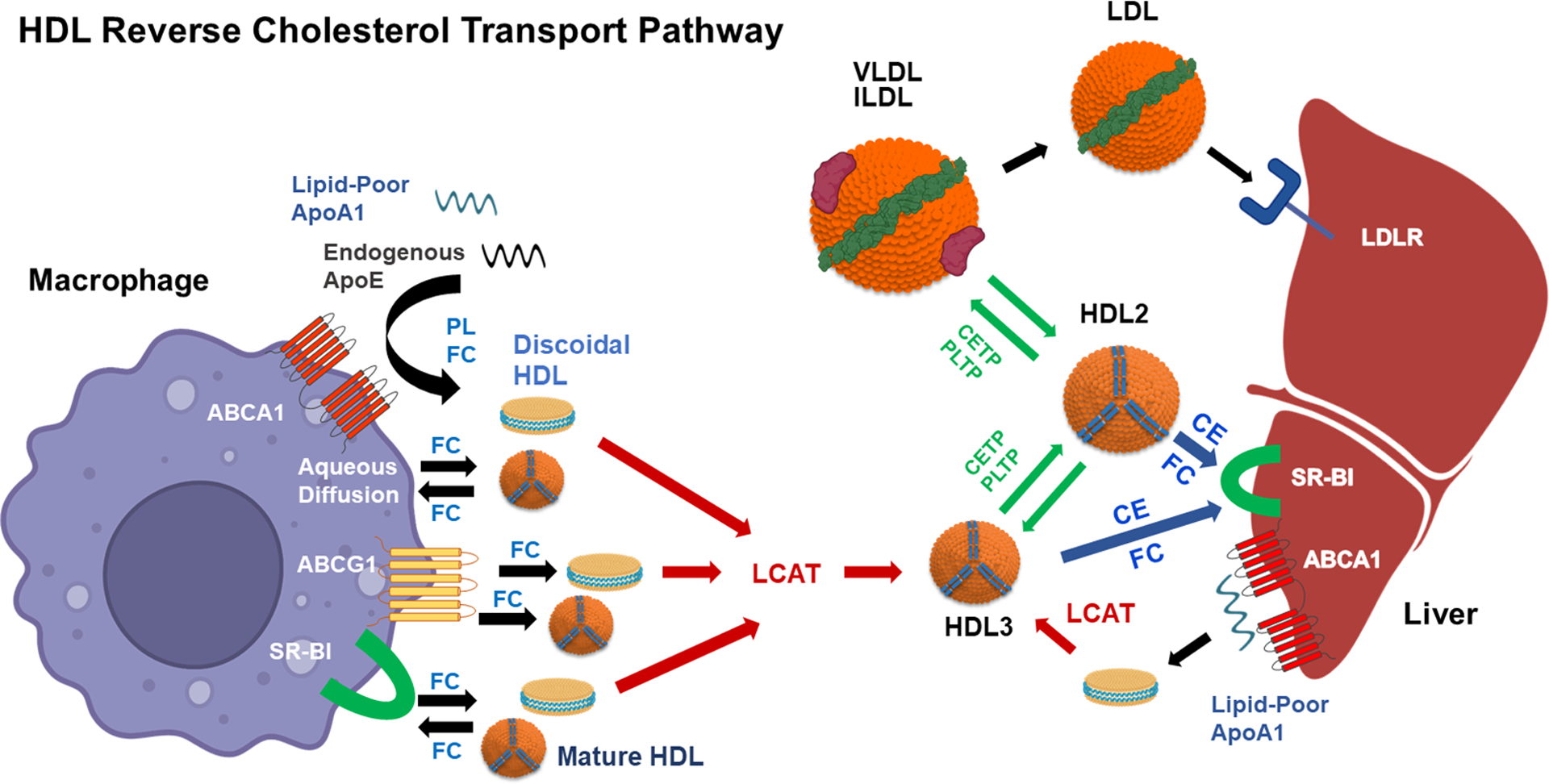
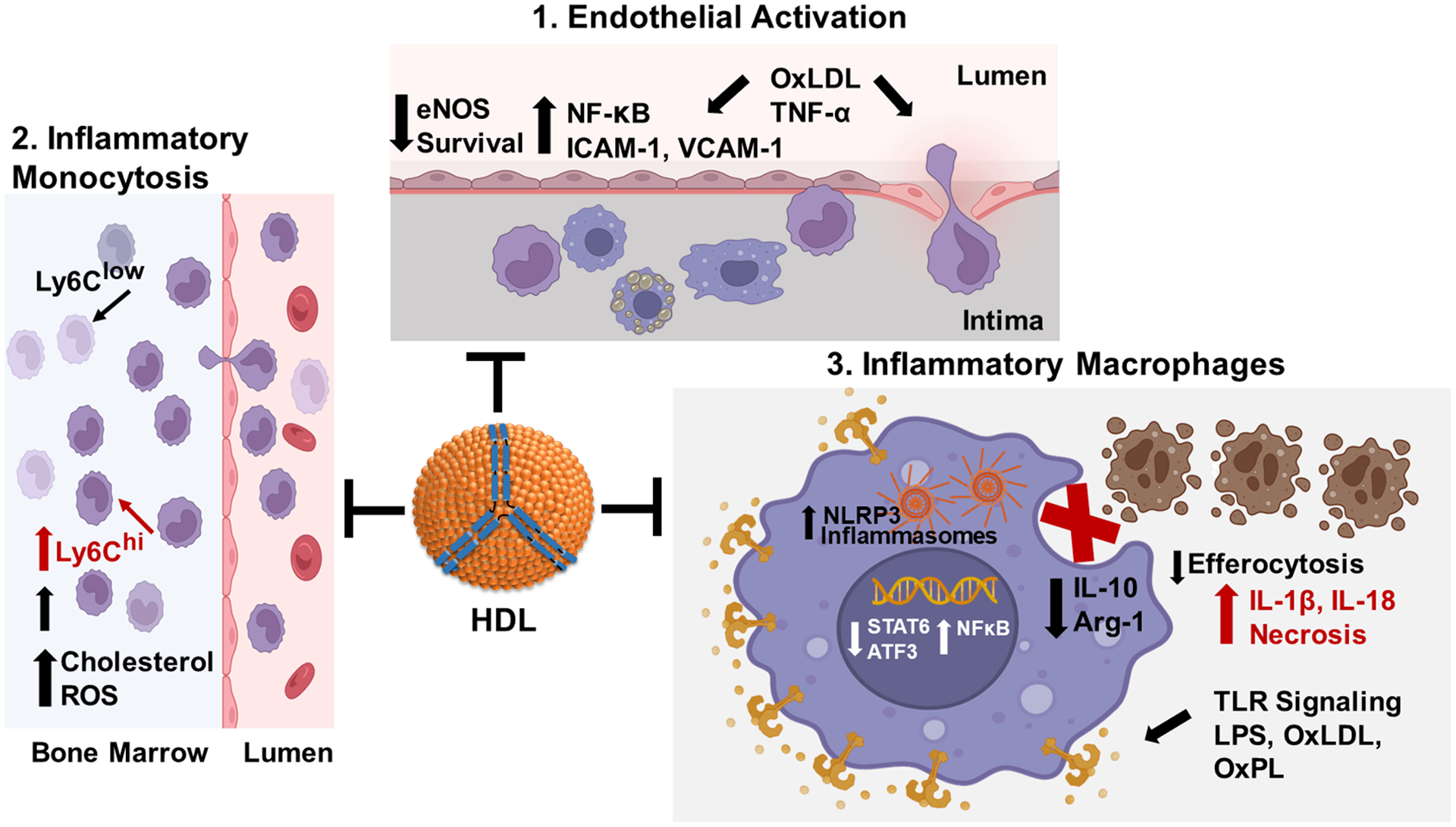
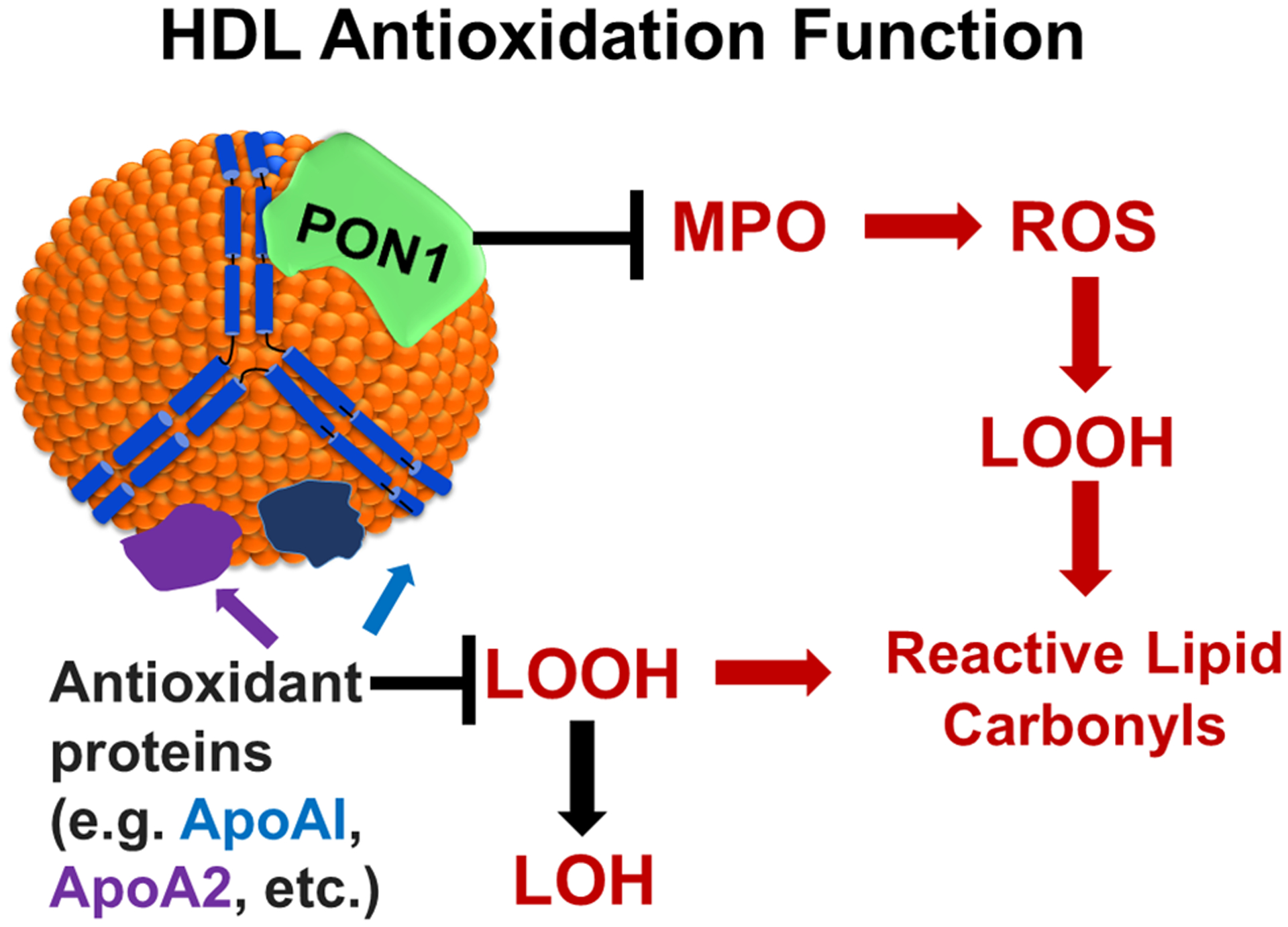
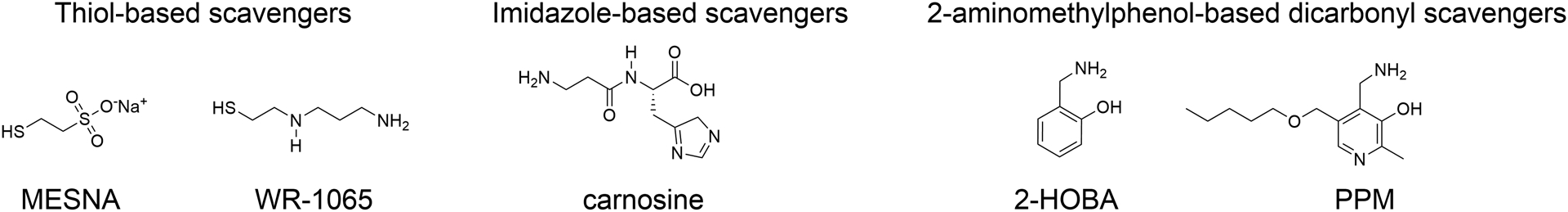
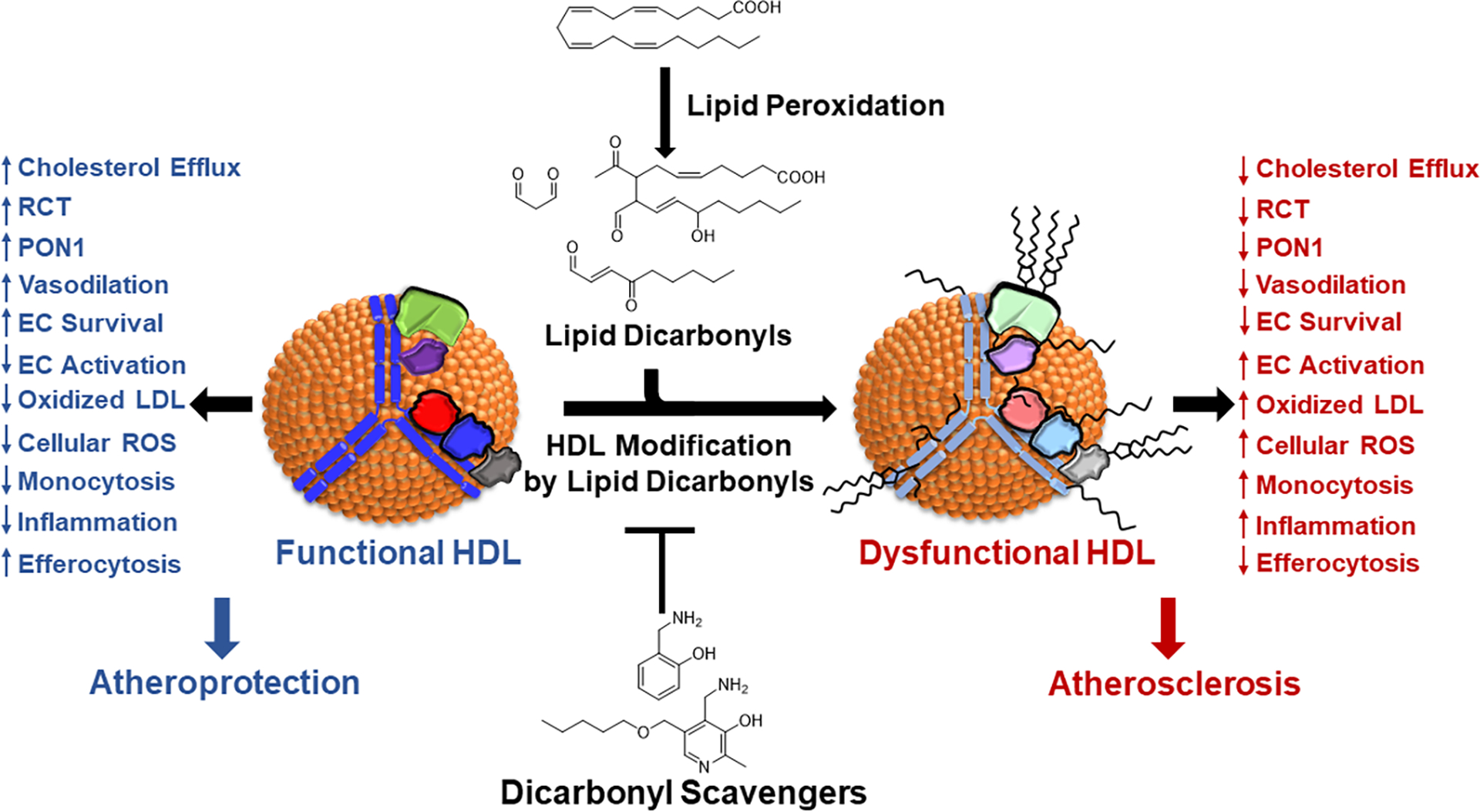
Epidemiologic studies detected an inverse relationship between HDL (high-density lipoprotein) cholesterol (HDL-C) levels and atherosclerotic cardiovascular disease (ASCVD), identifying HDL-C as a major risk factor for ASCVD and suggesting atheroprotective functions of HDL. However, the role of HDL-C as a mediator of risk for ASCVD has been called into question by the failure of HDL-C-raising drugs to reduce cardiovascular events in clinical trials. Progress in understanding the heterogeneous nature of HDL particles in terms of their protein, lipid, and small RNA composition has contributed to the realization that HDL-C levels do not necessarily reflect HDL function. The most examined atheroprotective function of HDL is reverse cholesterol transport, whereby HDL removes cholesterol from plaque macrophage foam cells and delivers it to the liver for processing and excretion into bile. Indeed, in several studies, HDL has shown inverse associations between HDL cholesterol efflux capacity and ASCVD in humans. Inflammation plays a key role in the pathogenesis of atherosclerosis and vulnerable plaque formation, and a fundamental function of HDL is suppression of inflammatory signaling in macrophages and other cells. Oxidation is also a critical process to ASCVD in promoting atherogenic oxidative modifications of LDL (low-density lipoprotein) and cellular inflammation. HDL and its proteins including apoAI (apolipoprotein AI) and PON1 (paraoxonase 1) prevent cellular oxidative stress and LDL modifications. Importantly, HDL in humans with ASCVD is oxidatively modified rendering HDL dysfunctional and proinflammatory. Modification of HDL with reactive carbonyl species, such as malondialdehyde and isolevuglandins, dramatically impairs the antiatherogenic functions of HDL. Importantly, treatment of murine models of atherosclerosis with scavengers of reactive dicarbonyls improves HDL function and reduces systemic inflammation, atherosclerosis development, and features of plaque instability. Here, we discuss the HDL antiatherogenic functions in relation to oxidative modifications and the potential of reactive dicarbonyl scavengers as a therapeutic approach for ASCVD.
Keywords: atherosclerosis; lipoproteins, HDL; macrophages; malondialdehyde; peroxidase.
Conflict of interest statement
Figures
References
-
- Gordon T, Castelli WP, Hjortland MC, Kannel WB and Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–14. - PubMed
-
- Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR Jr., Bangdiwala S and Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. - PubMed
-
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D and Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. - PMC - PubMed
-
- Karjalainen MK, Holmes MV, Wang Q, Anufrieva O, Kahonen M, Lehtimaki T, Havulinna AS, Kristiansson K, Salomaa V, Perola M, Viikari JS, Raitakari OT, Jarvelin MR, Ala-Korpela M and Kettunen J. Apolipoprotein A-I concentrations and risk of coronary artery disease: A Mendelian randomization study. Atherosclerosis. 2020;299:56–63. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
