

Plasma virome dynamics in chronic hepatitis B virus infected patients
- PMID: 37228370
- PMCID: PMC10203228
- DOI: 10.3389/fmicb.2023.1172574
Plasma virome dynamics in chronic hepatitis B virus infected patients
Abstract
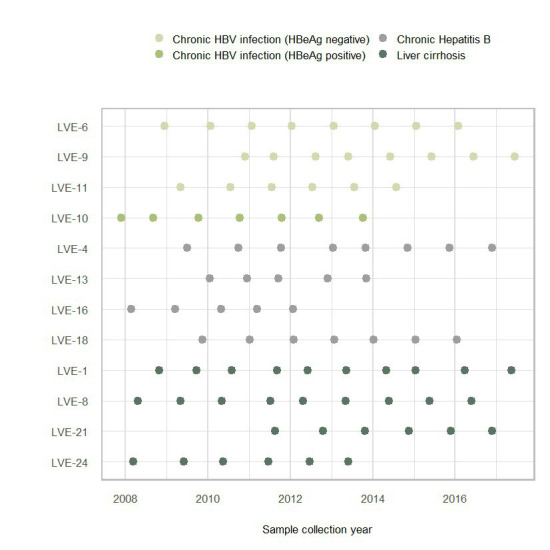
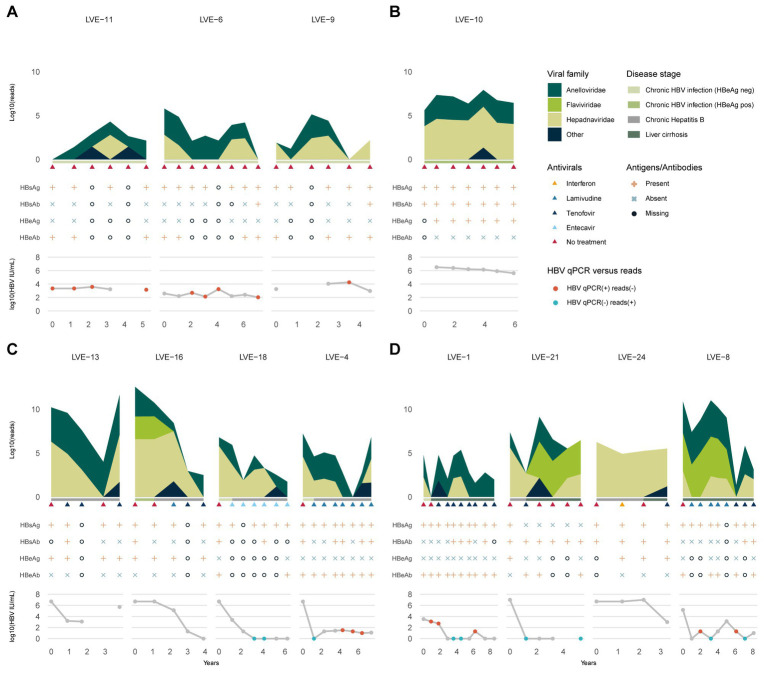
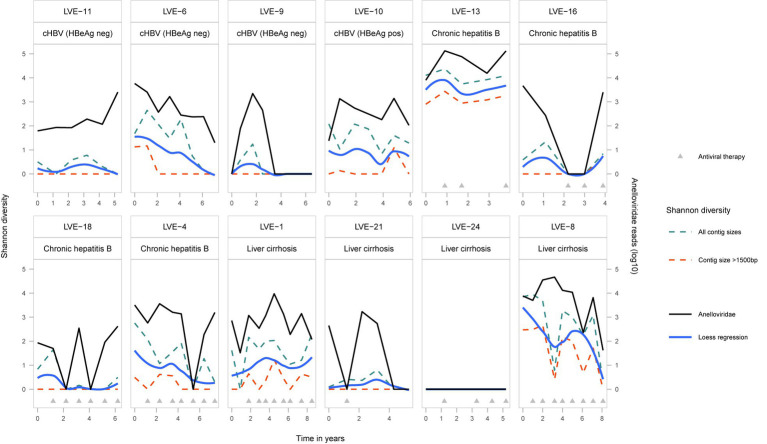
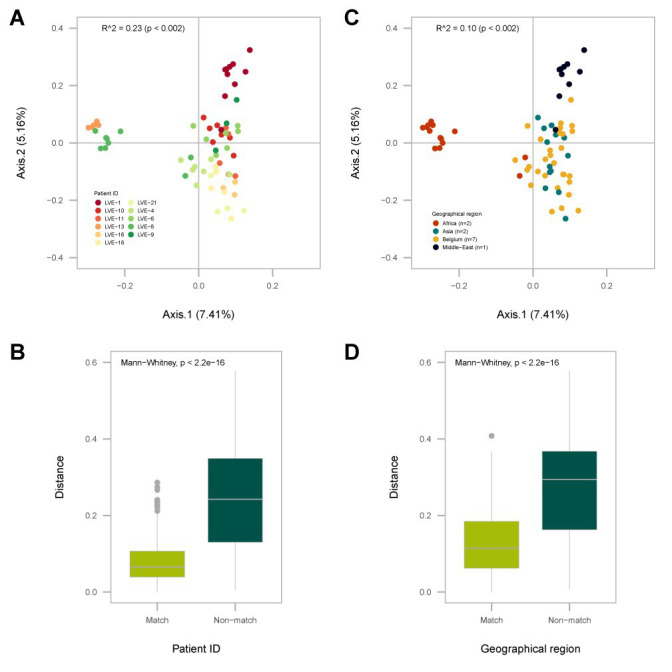
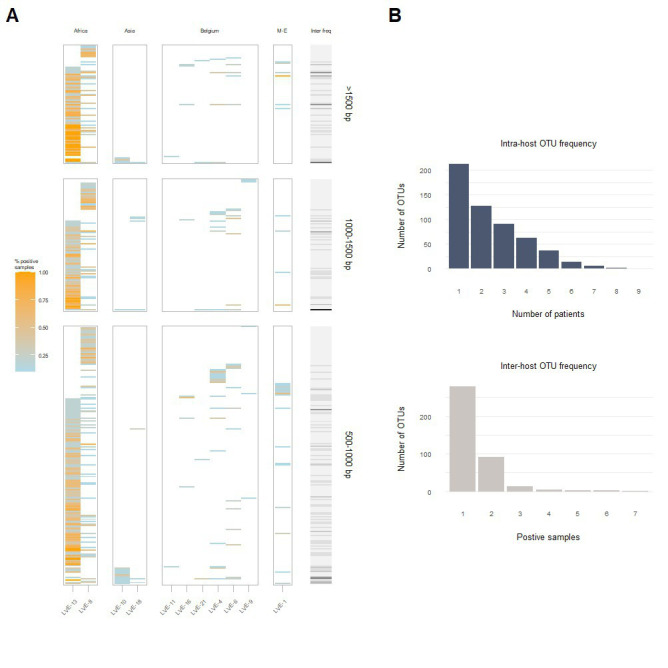
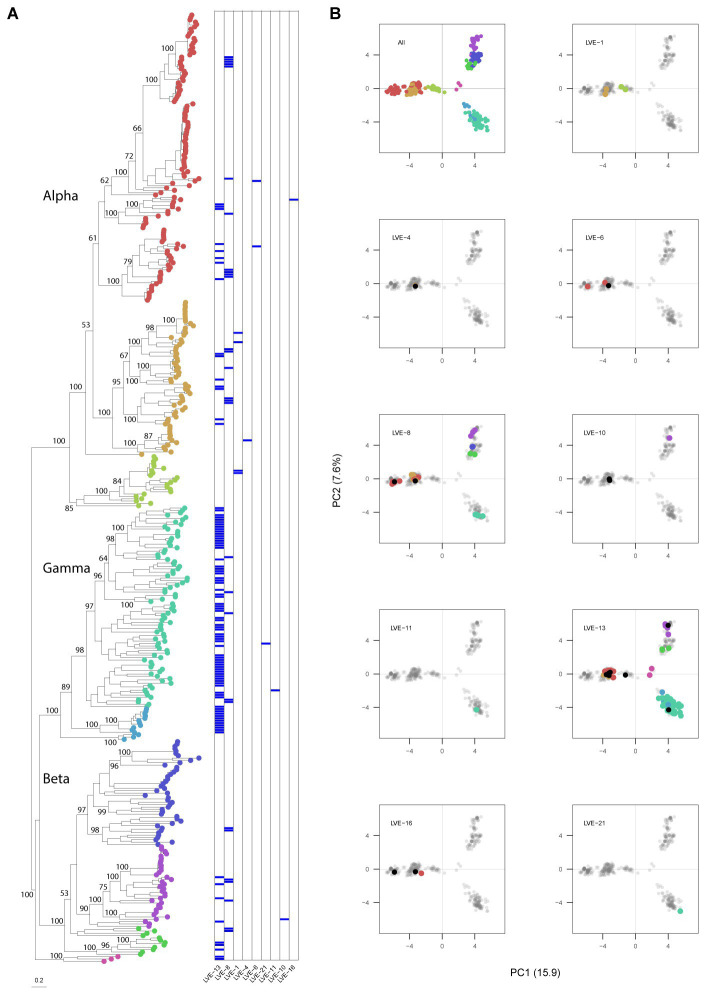
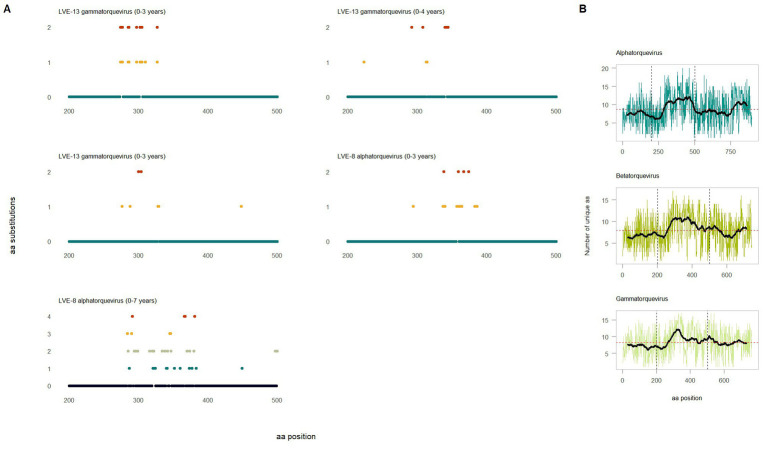
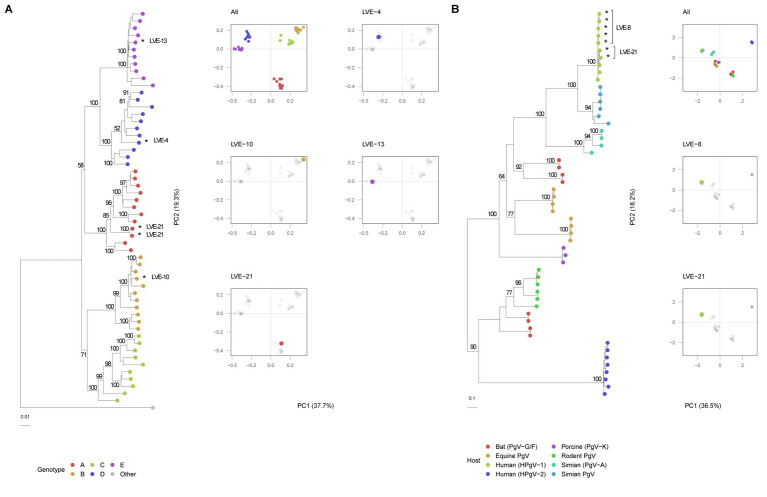
The virome remains an understudied domain of the human microbiome. The role of commensal viruses on the outcome of infections with known pathogens is not well characterized. In this study we aimed to characterize the longitudinal plasma virome dynamics in chronic hepatitis B virus (HBV) infected patients. Eighty-five longitudinal plasma samples were collected from 12 chronic HBV infected individuals that were classified in the four stages of HBV infection. The virome was characterized with an optimized viral extraction protocol and deep-sequenced on a NextSeq 2500 platform. The plasma virome was primarily composed of members of the Anello- Flavi-, and Hepadnaviridae (HBV) families. The virome structure and dynamics did not correlate with the different stages of chronic HBV infection nor with the administration of antiviral therapy. We observed a higher intrapersonal similarity of viral contigs. Genomic analysis of viruses observed in multiple timepoint demonstrated the presence of a dynamic community. This study comprehensively assessed the blood virome structure in chronic HBV infected individuals and provided insights in the longitudinal development of this viral community.
Keywords: anellovirus; blood; hepatitis B virus; metagenomic; pegivirus; plasma; virome.
Copyright © 2023 Thijssen, Tacke, Van Espen, Cassiman, Naser Aldine, Nevens, Van Ranst, Matthijnssens and Pourkarim.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources