

Single-nucleus RNA sequencing of pre-malignant liver reveals disease-associated hepatocyte state with HCC prognostic potential
- PMID: 37228755
- PMCID: PMC10203275
- DOI: 10.1016/j.xgen.2023.100301
Single-nucleus RNA sequencing of pre-malignant liver reveals disease-associated hepatocyte state with HCC prognostic potential
Abstract
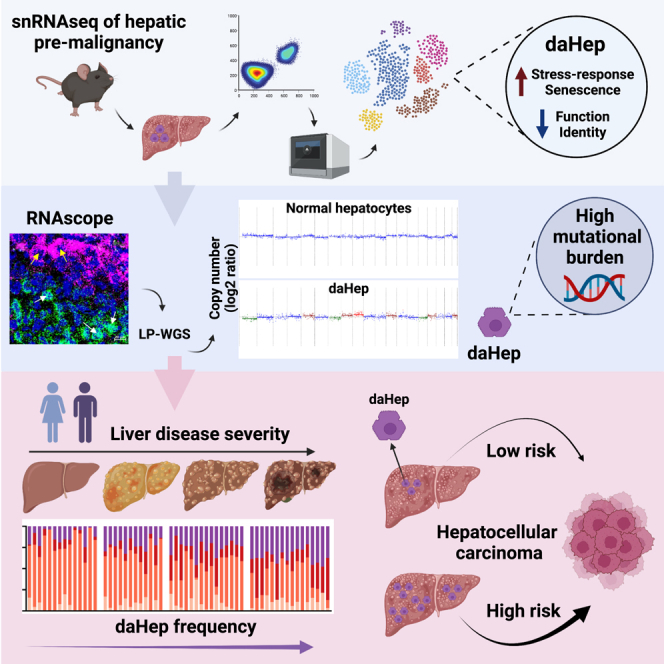
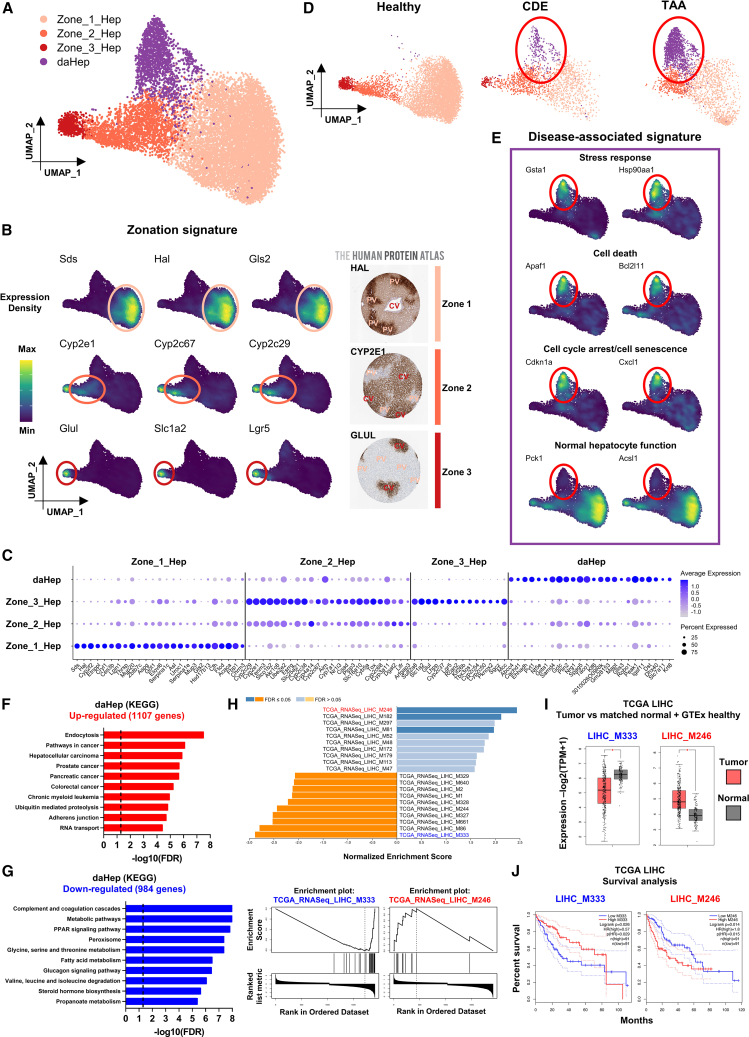
Current approaches to staging chronic liver diseases have limited utility for predicting liver cancer risk. Here, we employed single-nucleus RNA sequencing (snRNA-seq) to characterize the cellular microenvironment of healthy and pre-malignant livers using two distinct mouse models. Downstream analyses unraveled a previously uncharacterized disease-associated hepatocyte (daHep) transcriptional state. These cells were absent in healthy livers but increasingly prevalent as chronic liver disease progressed. Copy number variation (CNV) analysis of microdissected tissue demonstrated that daHep-enriched regions are riddled with structural variants, suggesting these cells represent a pre-malignant intermediary. Integrated analysis of three recent human snRNA-seq datasets confirmed the presence of a similar phenotype in human chronic liver disease and further supported its enhanced mutational burden. Importantly, we show that high daHep levels precede carcinogenesis and predict a higher risk of hepatocellular carcinoma development. These findings may change the way chronic liver disease patients are staged, surveilled, and risk stratified.
Keywords: HCC; cancer surveillance; chronic liver disease; daHep; disease-associated hepatocyte; hepatocellular carcinoma; pre-malignancy; prognostic biomarker; snRNA-seq.
© 2023 The Author(s).
Conflict of interest statement
M.A.F. is the founder and shareholder of Celesta Therapeutics.
Figures






References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
