

Integrating genetics with single-cell multiomic measurements across disease states identifies mechanisms of beta cell dysfunction in type 2 diabetes
- PMID: 37231096
- PMCID: PMC10550816
- DOI: 10.1038/s41588-023-01397-9
Integrating genetics with single-cell multiomic measurements across disease states identifies mechanisms of beta cell dysfunction in type 2 diabetes
Abstract
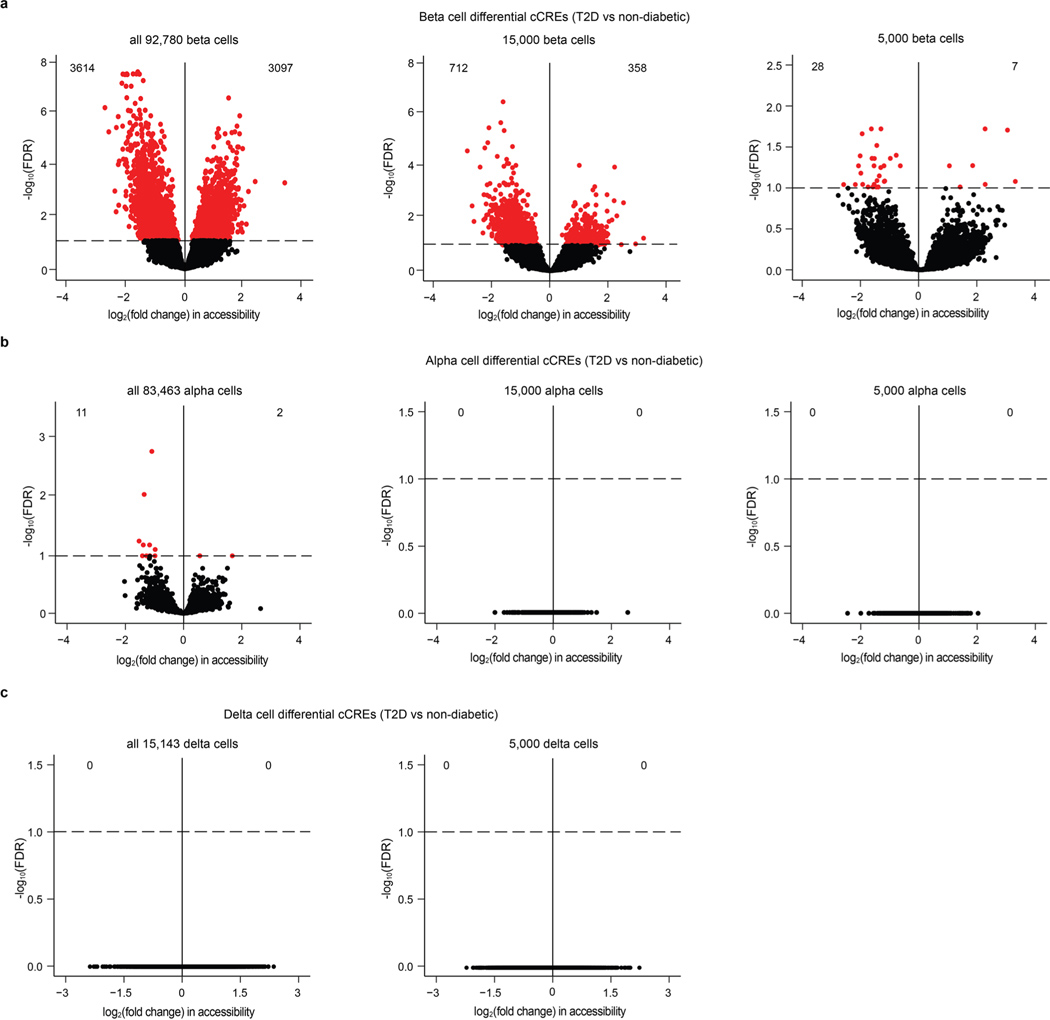
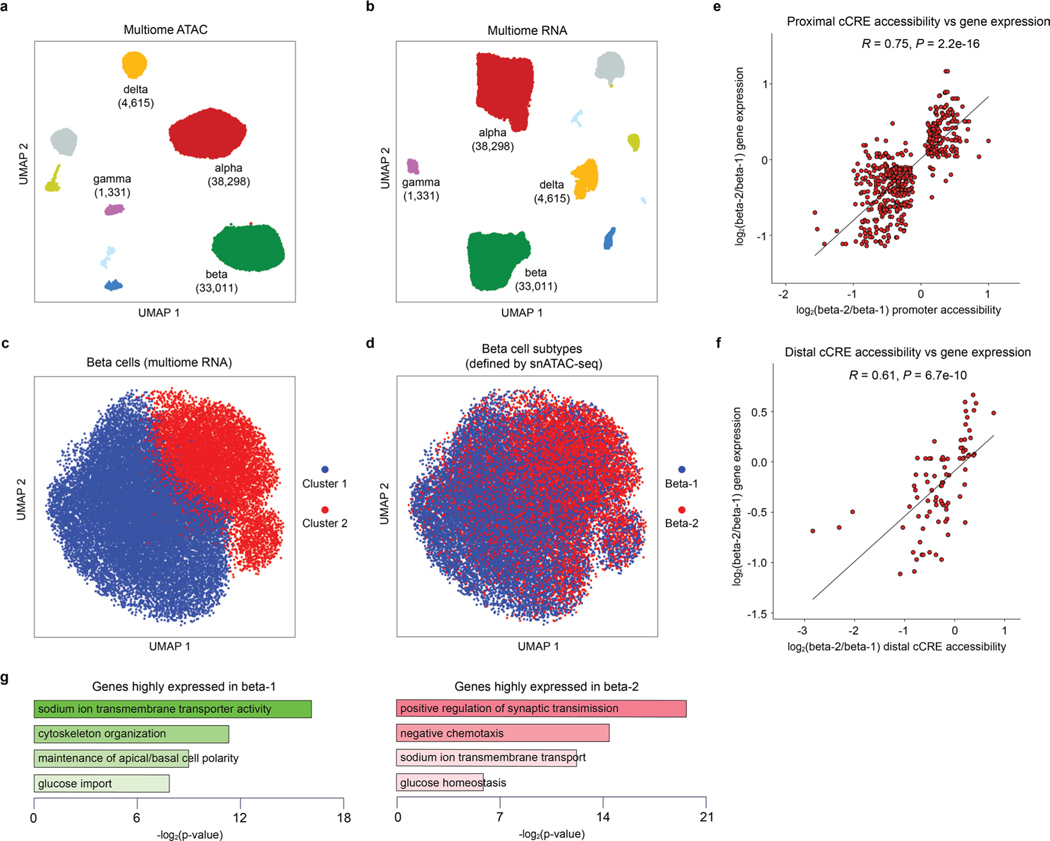
Dysfunctional pancreatic islet beta cells are a hallmark of type 2 diabetes (T2D), but a comprehensive understanding of the underlying mechanisms, including gene dysregulation, is lacking. Here we integrate information from measurements of chromatin accessibility, gene expression and function in single beta cells with genetic association data to nominate disease-causal gene regulatory changes in T2D. Using machine learning on chromatin accessibility data from 34 nondiabetic, pre-T2D and T2D donors, we identify two transcriptionally and functionally distinct beta cell subtypes that undergo an abundance shift during T2D progression. Subtype-defining accessible chromatin is enriched for T2D risk variants, suggesting a causal contribution of subtype identity to T2D. Both beta cell subtypes exhibit activation of a stress-response transcriptional program and functional impairment in T2D, which is probably induced by the T2D-associated metabolic environment. Our findings demonstrate the power of multimodal single-cell measurements combined with machine learning for characterizing mechanisms of complex diseases.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
K.J.G. holds stock in Vertex Pharmaceuticals and Neurocrine Biosciences. J.C. is now employed by and holds stock in Pfizer Inc. The other authors declare no competing interests.
Figures














Update of
-
Integration of single-cell multiomic measurements across disease states with genetics identifies mechanisms of beta cell dysfunction in type 2 diabetes.bioRxiv [Preprint]. 2023 Jan 2:2022.12.31.522386. doi: 10.1101/2022.12.31.522386. bioRxiv. 2023. Update in: Nat Genet. 2023 Jun;55(6):984-994. doi: 10.1038/s41588-023-01397-9. PMID: 36711922 Free PMC article. Updated. Preprint.
References
-
- Wojtusciszyn A, Armanet M, Morel P, Berney T. & Bosco D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 51, 1843–1852 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK105541/DK/NIDDK NIH HHS/United States
- R01 DK114650/DK/NIDDK NIH HHS/United States
- R01 DK068471/DK/NIDDK NIH HHS/United States
- DP3 DK112155/DK/NIDDK NIH HHS/United States
- R01 DK122607/DK/NIDDK NIH HHS/United States
- UC4 DK112232/DK/NIDDK NIH HHS/United States
- S10 OD026929/OD/NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
- P30 DK116074/DK/NIDDK NIH HHS/United States
- U01 DK123716/DK/NIDDK NIH HHS/United States
- U01 DK120447/DK/NIDDK NIH HHS/United States
- UC4 DK112217/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
