

AIomics: Exploring More of the Proteome Using Mass Spectral Libraries Extended by Artificial Intelligence
- PMID: 37232537
- PMCID: PMC10542943
- DOI: 10.1021/acs.jproteome.2c00807
AIomics: Exploring More of the Proteome Using Mass Spectral Libraries Extended by Artificial Intelligence
Abstract
The unbounded permutations of biological molecules, including proteins and their constituent peptides, present a dilemma in identifying the components of complex biosamples. Sequence search algorithms used to identify peptide spectra can be expanded to cover larger classes of molecules, including more modifications, isoforms, and atypical cleavage, but at the cost of false positives or false negatives due to the simplified spectra they compute from sequence records. Spectral library searching can help solve this issue by precisely matching experimental spectra to library spectra with excellent sensitivity and specificity. However, compiling spectral libraries that span entire proteomes is pragmatically difficult. Neural networks that predict complete spectra containing a full range of annotated and unannotated ions can be used to replace these simplified spectra with libraries of fully predicted spectra, including modified peptides. Using such a network, we created predicted spectral libraries that were used to rescore matches from a sequence search done over a large search space, including a large number of modifications. Rescoring improved the separation of true and false hits by 82%, yielding an 8% increase in peptide identifications, including a 21% increase in nonspecifically cleaved peptides and a 17% increase in phosphopeptides.
Keywords: algorithms; machine learning; peptides; proteome analysis; search engine methods; tandem mass spectrometry.
Conflict of interest statement
The authors declare no competing financial interests. All commercial instruments, software, and materials used in the study are for experimental purposes only. Such identification does not intend recommendation or endorsement by the National Institute of Standards and Technology, nor does it intend that the materials, software, or instruments used are necessarily the best available for the purpose.
Figures

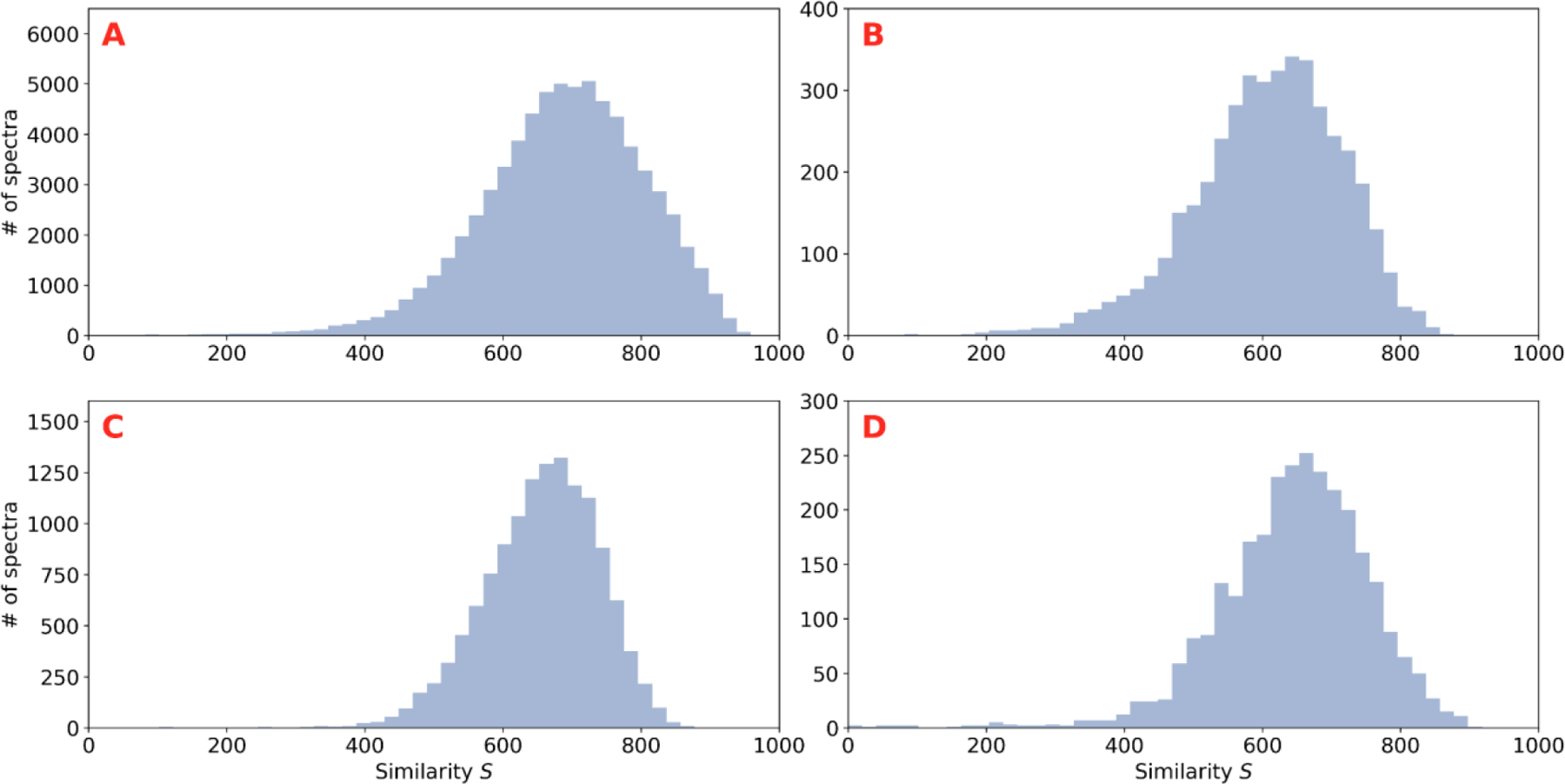


References
-
- Shilov IV; Seymour SL; Patel AA; Loboda A; Tang WH; Keating SP; Hunter CL; Nuwaysir LM; Schaeffer DA The Paragon Algorithm, a Next Generation Search Engine That Uses Sequence Temperature Values and Feature Probabilities to Identify Peptides from Tandem Mass Spectra. Mol. Cell. Proteomics 2007, 6 (9), 1638–1655. 10.1074/mcp.T600050-MCP200. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
