

Genome-scale functional genomics identify genes preferentially essential for multiple myeloma cells compared to other neoplasias
- PMID: 37237081
- PMCID: PMC10918623
- DOI: 10.1038/s43018-023-00550-x
Genome-scale functional genomics identify genes preferentially essential for multiple myeloma cells compared to other neoplasias
Abstract
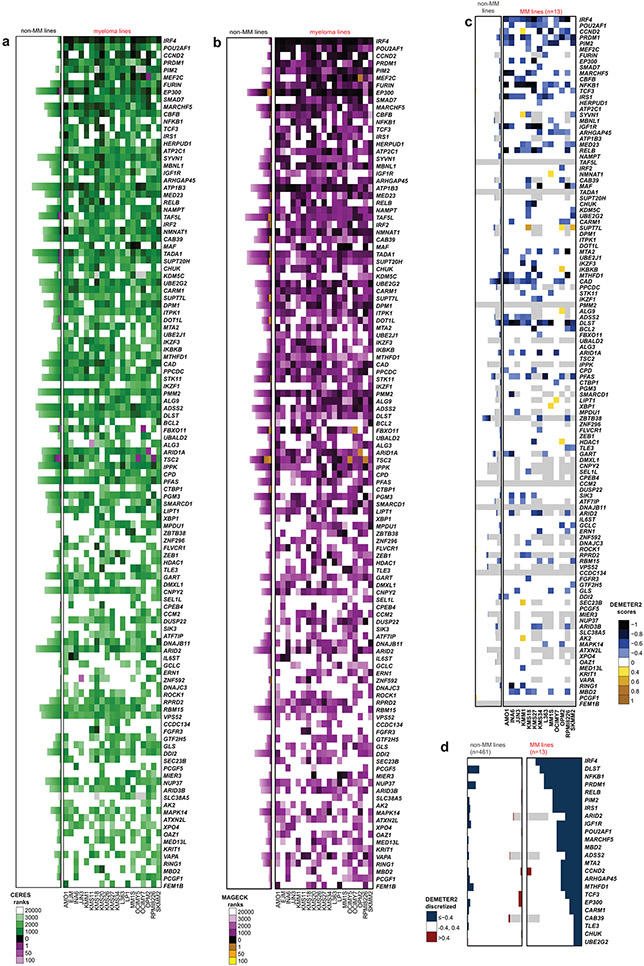
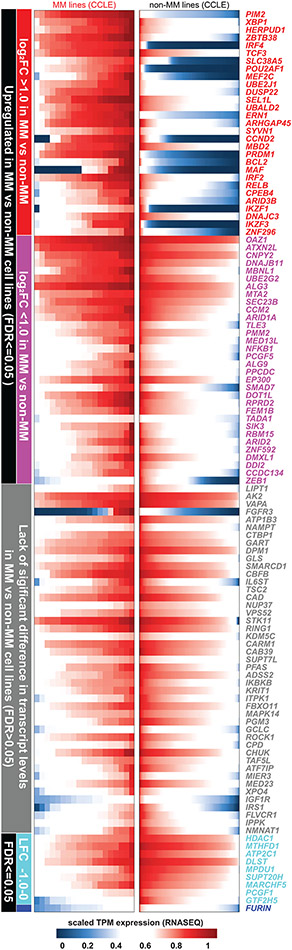
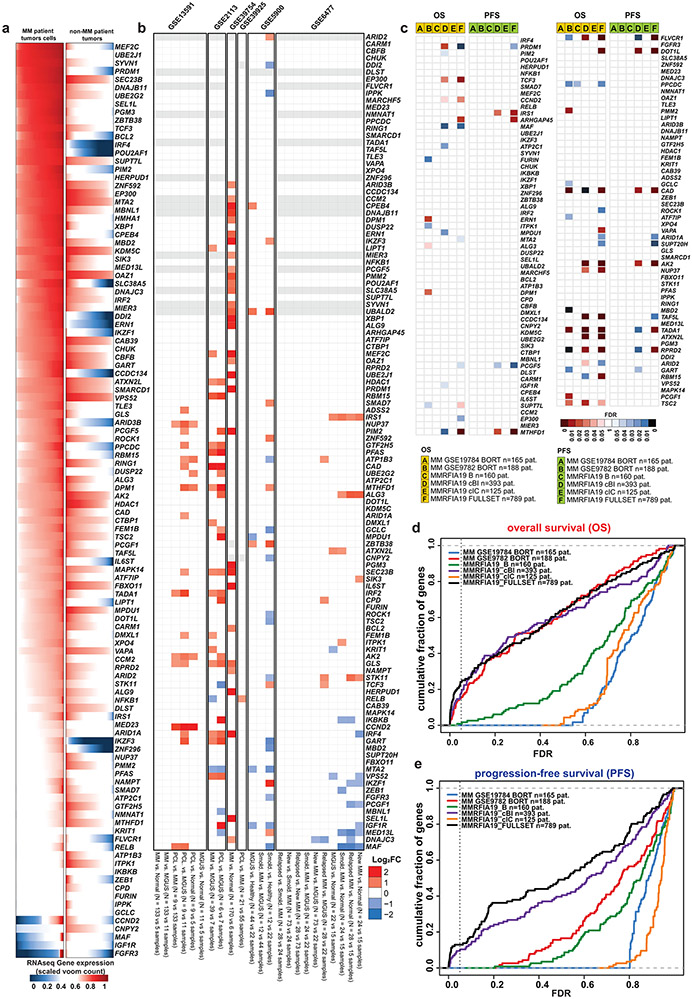
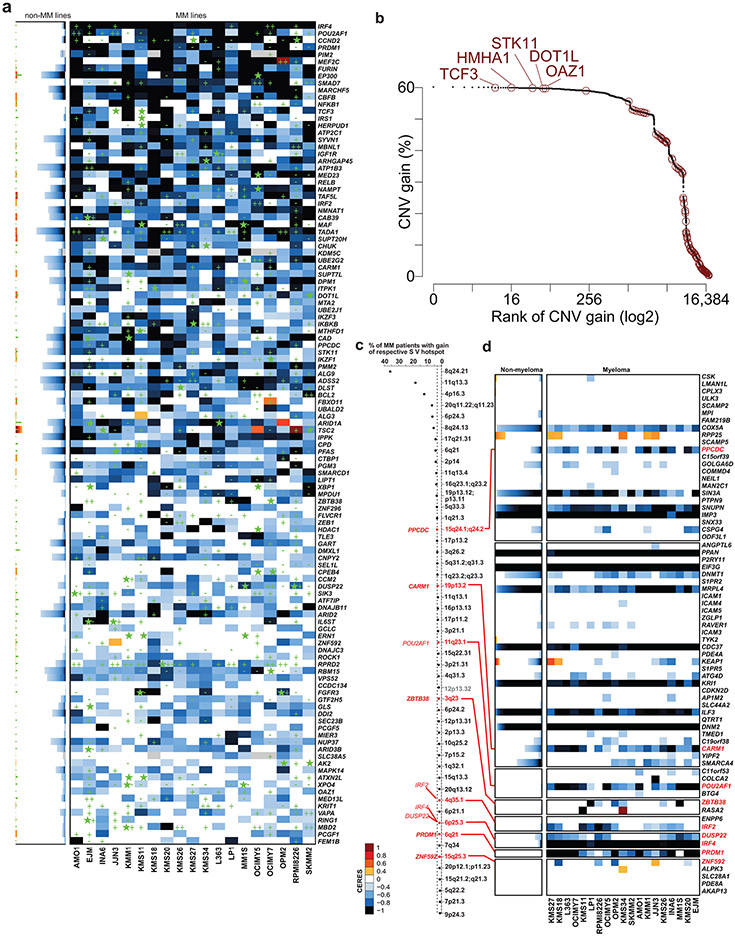
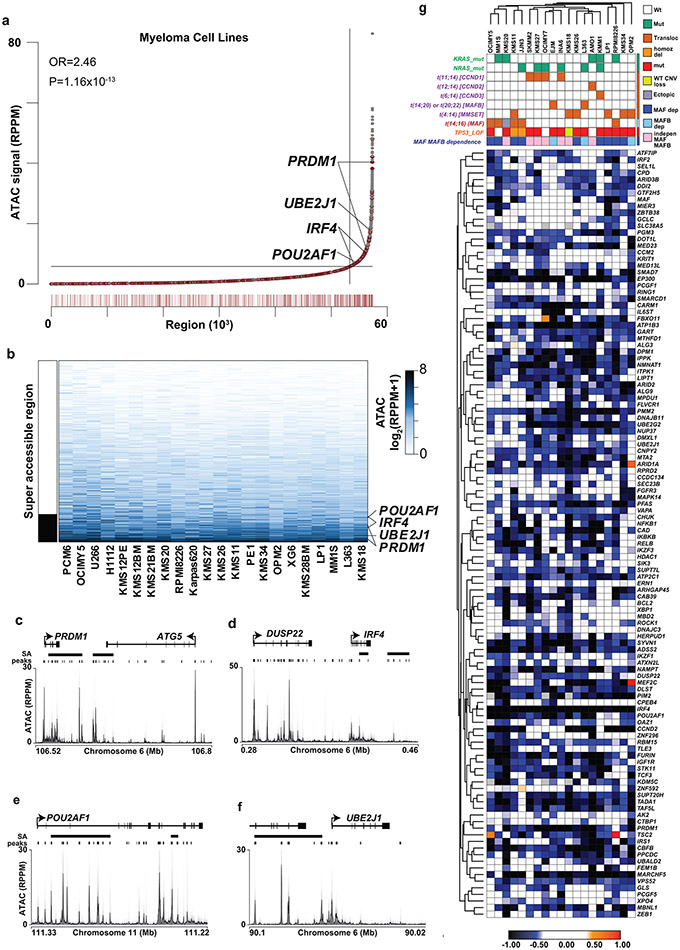
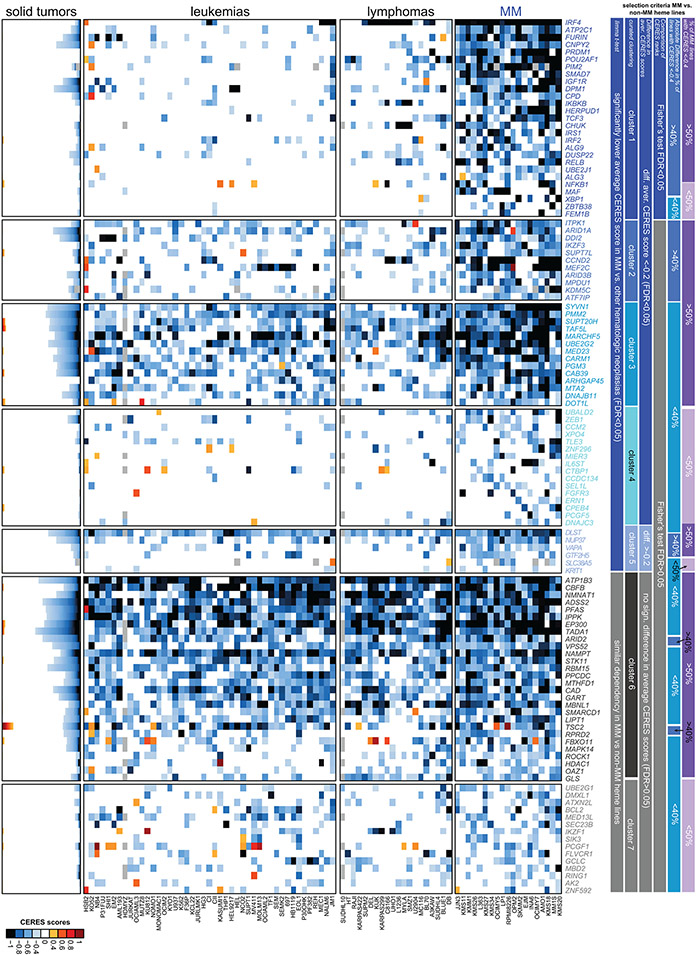
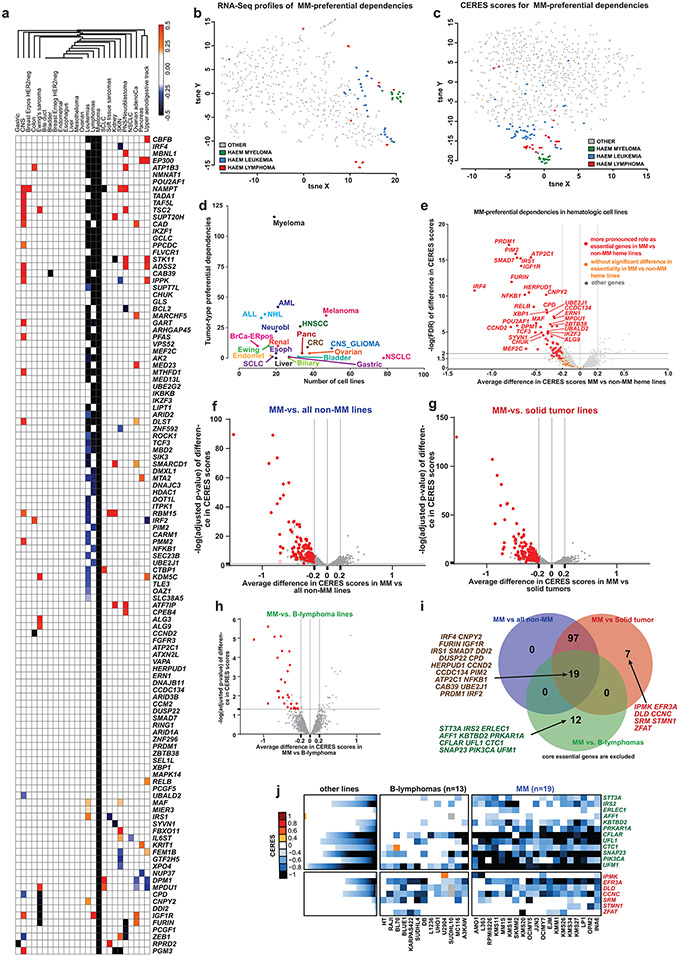
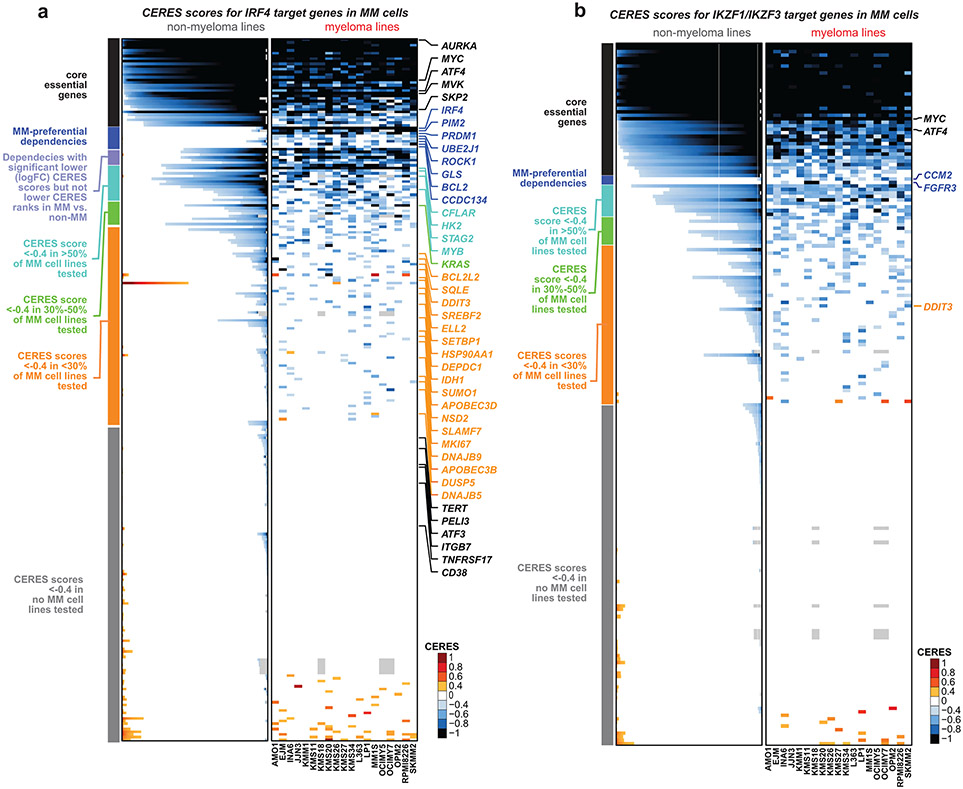
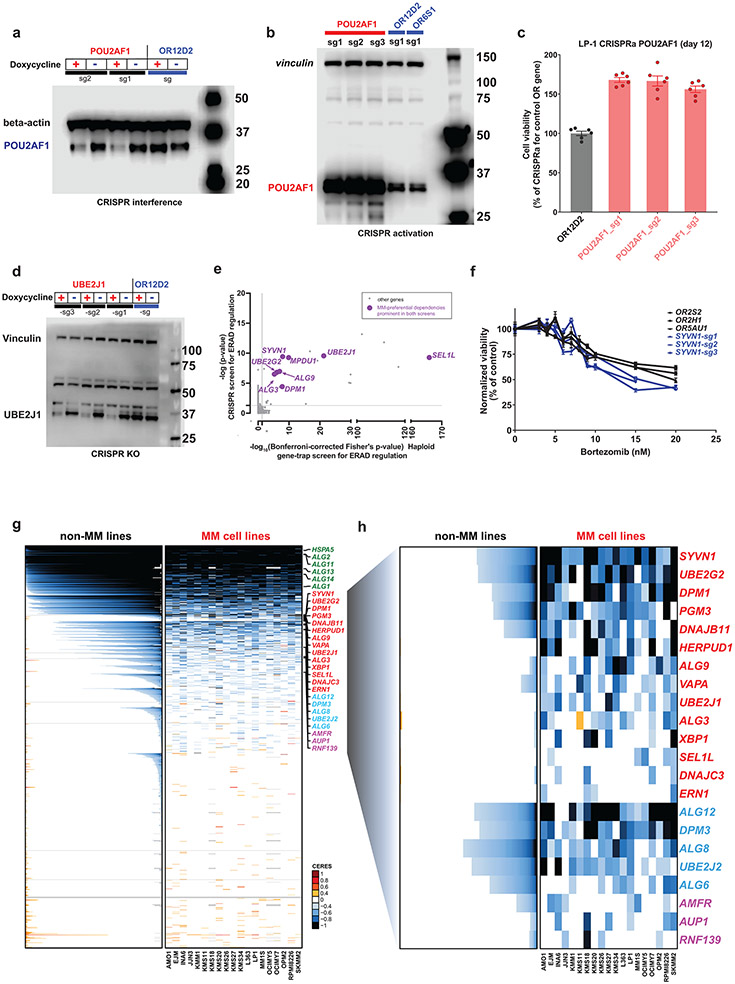
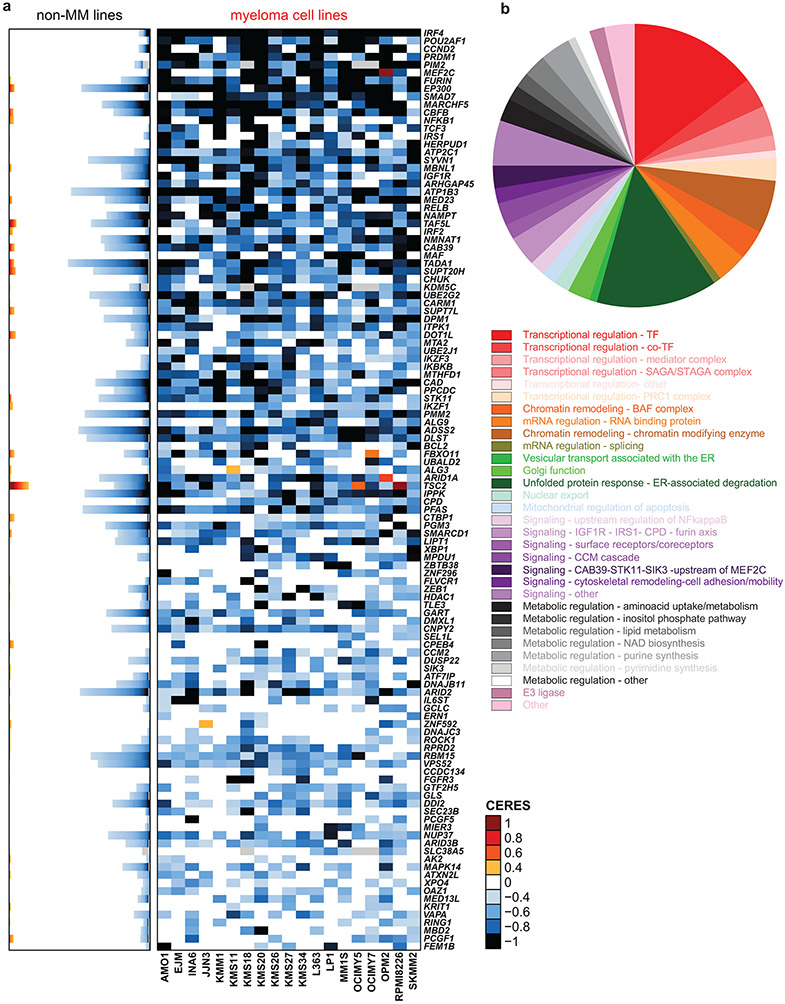
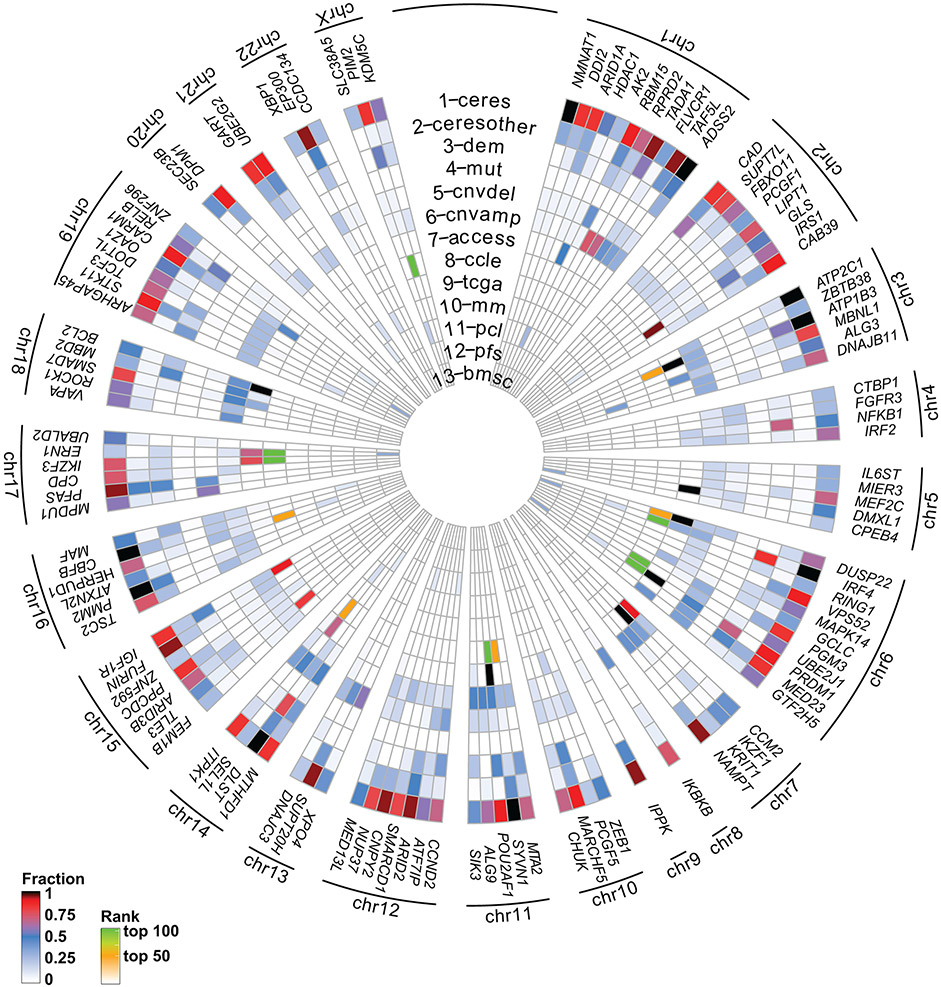
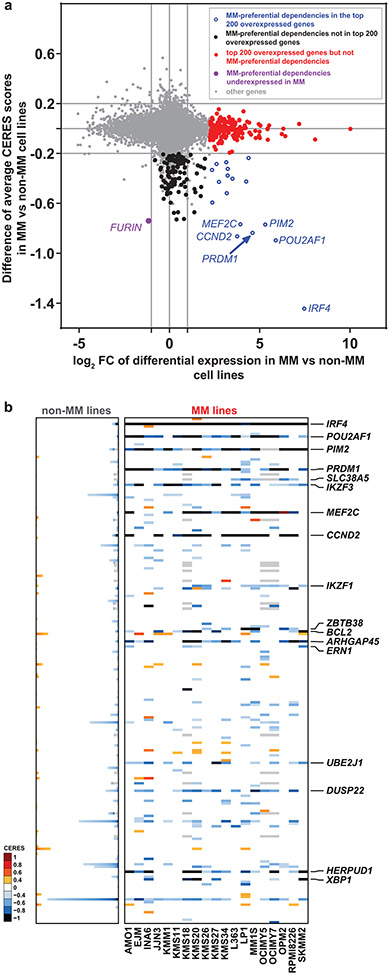
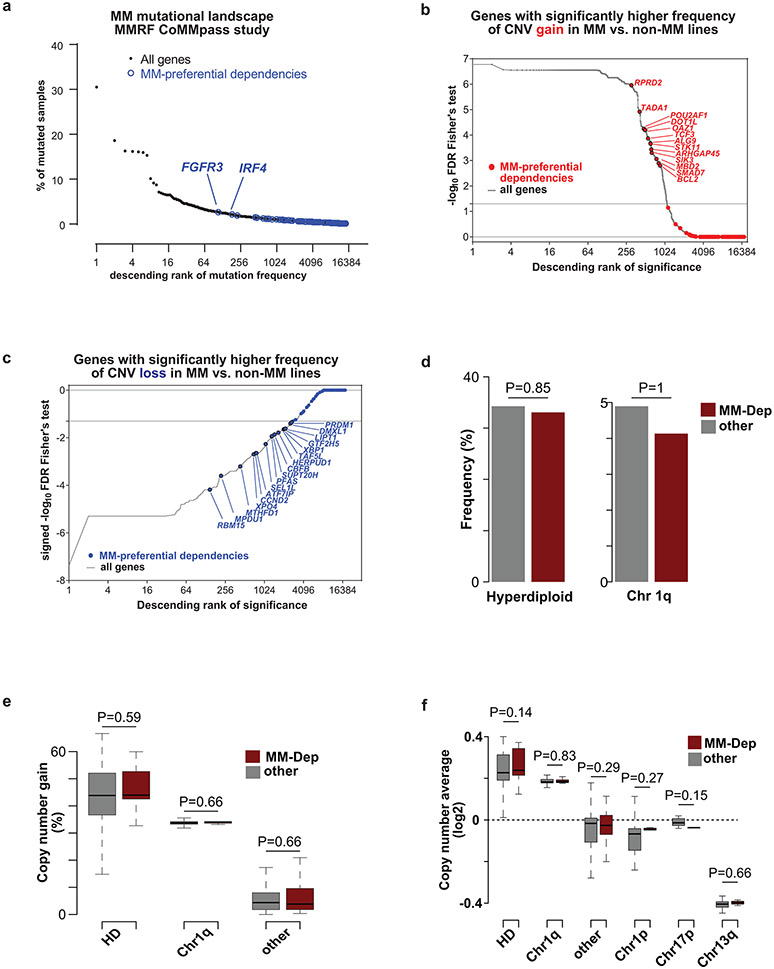
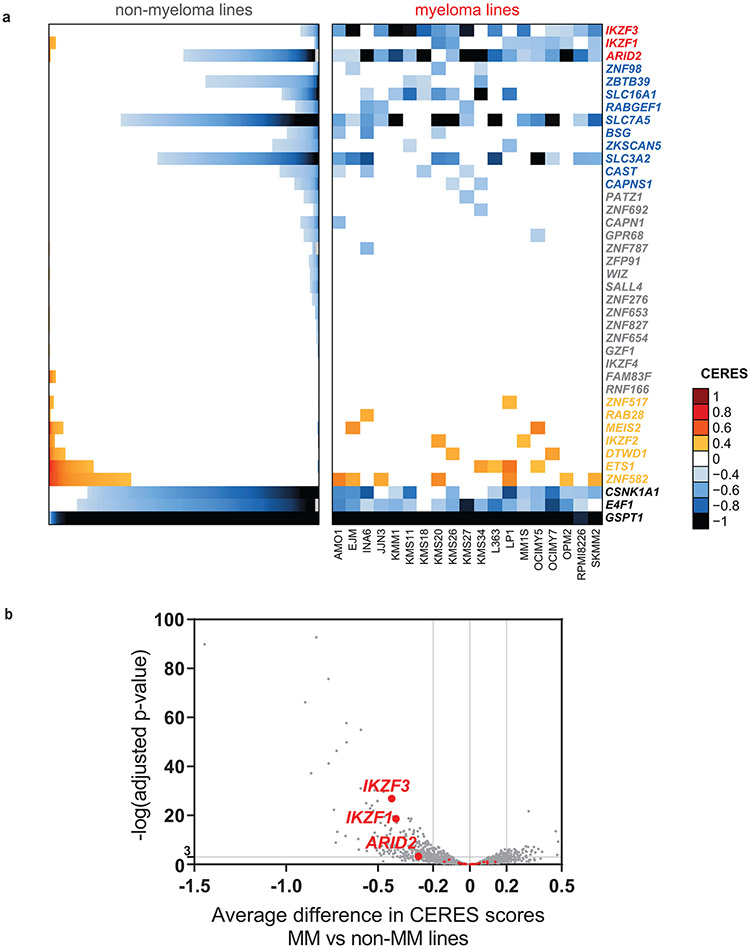
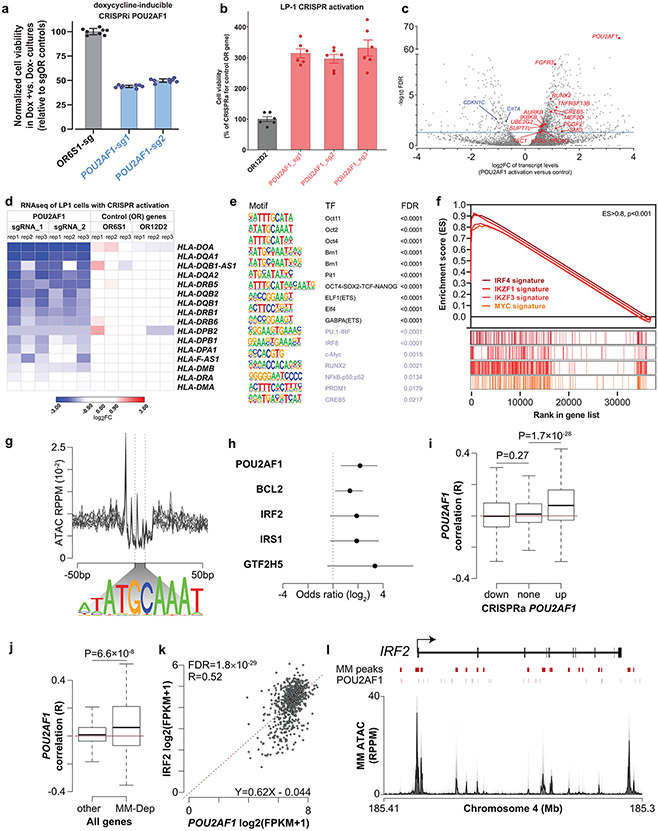
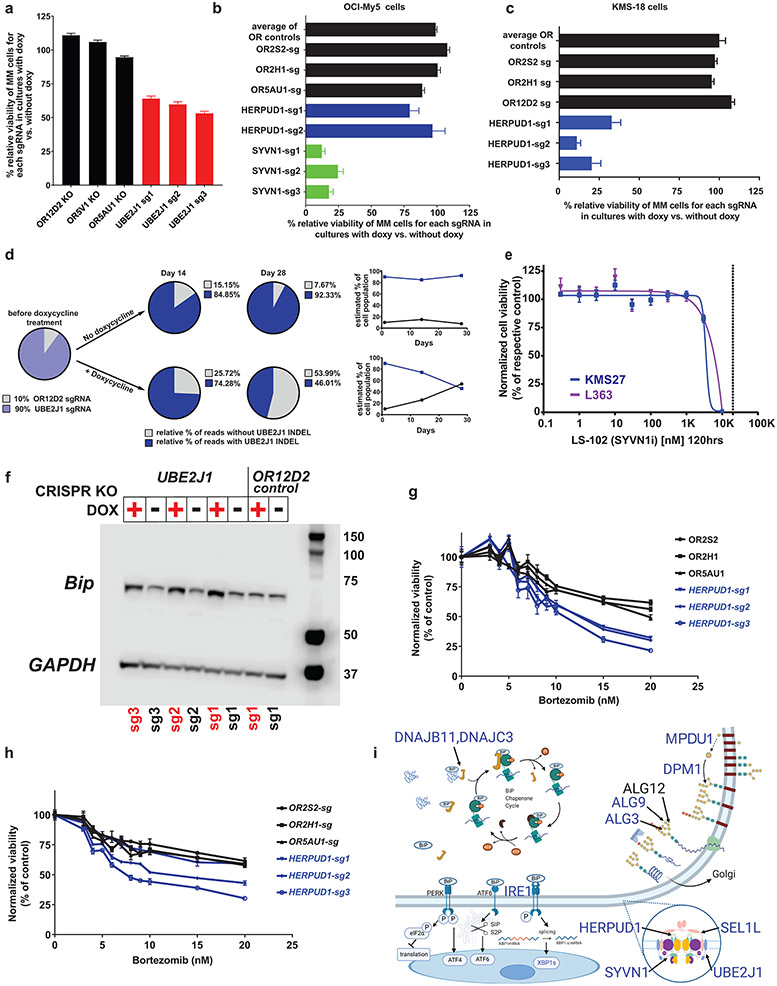
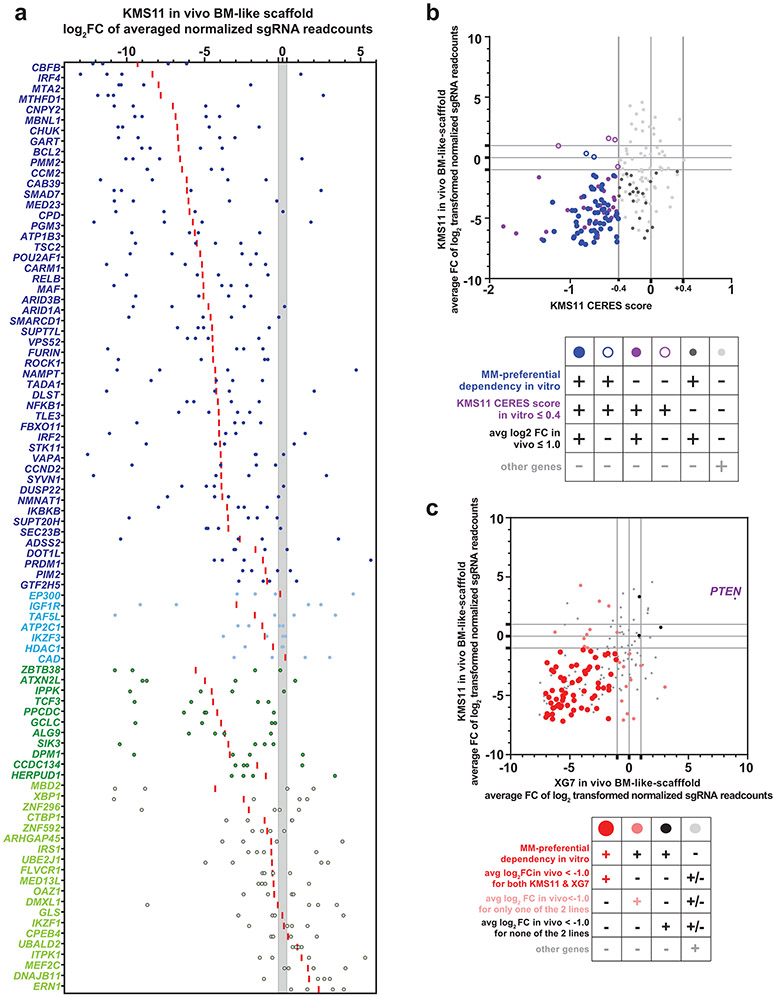
Clinical progress in multiple myeloma (MM), an incurable plasma cell (PC) neoplasia, has been driven by therapies that have limited applications beyond MM/PC neoplasias and do not target specific oncogenic mutations in MM. Instead, these agents target pathways critical for PC biology yet largely dispensable for malignant or normal cells of most other lineages. Here we systematically characterized the lineage-preferential molecular dependencies of MM through genome-scale clustered regularly interspaced short palindromic repeats (CRISPR) studies in 19 MM versus hundreds of non-MM lines and identified 116 genes whose disruption more significantly affects MM cell fitness compared with other malignancies. These genes, some known, others not previously linked to MM, encode transcription factors, chromatin modifiers, endoplasmic reticulum components, metabolic regulators or signaling molecules. Most of these genes are not among the top amplified, overexpressed or mutated in MM. Functional genomics approaches thus define new therapeutic targets in MM not readily identifiable by standard genomic, transcriptional or epigenetic profiling analyses.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures

References
-
- Andrulis M. et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov 3, 862–9 (2013). - PubMed
Methods-Only References
-
- Dempster JM et al. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv, 720243 (2019).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
