

Metabolic Myopathies in the Era of Next-Generation Sequencing
- PMID: 37239314
- PMCID: PMC10217901
- DOI: 10.3390/genes14050954
Metabolic Myopathies in the Era of Next-Generation Sequencing
Abstract
Metabolic myopathies are rare inherited disorders that deserve more attention from neurologists and pediatricians. Pompe disease and McArdle disease represent some of the most common diseases in clinical practice; however, other less common diseases are now better-known. In general the pathophysiology of metabolic myopathies needs to be better understood. Thanks to the advent of next-generation sequencing (NGS), genetic testing has replaced more invasive investigations and sophisticated enzymatic assays to reach a final diagnosis in many cases. The current diagnostic algorithms for metabolic myopathies have integrated this paradigm shift and restrict invasive investigations for complicated cases. Moreover, NGS contributes to the discovery of novel genes and proteins, providing new insights into muscle metabolism and pathophysiology. More importantly, a growing number of these conditions are amenable to therapeutic approaches such as diets of different kinds, exercise training protocols, and enzyme replacement therapy or gene therapy. Prevention and management-notably of rhabdomyolysis-are key to avoiding serious and potentially life-threatening complications and improving patients' quality of life. Although not devoid of limitations, the newborn screening programs that are currently mushrooming across the globe show that early intervention in metabolic myopathies is a key factor for better therapeutic efficacy and long-term prognosis. As a whole NGS has largely increased the diagnostic yield of metabolic myopathies, but more invasive but classical investigations are still critical when the genetic diagnosis is unclear or when it comes to optimizing the follow-up and care of these muscular disorders.
Keywords: Cori–Forbes disease; McArdle disease; Pompe disease; TK2 myopathy; Tarui disease; glycogen storage disorders; lipid storage diseases; metabolic myopathies; mitochondrial myopathies; muscle glycogenoses; muscle metabolism; polyglucosan body myopathies.
Conflict of interest statement
The authors have no conflicts of interest to disclose that might be related to this paper.
Figures
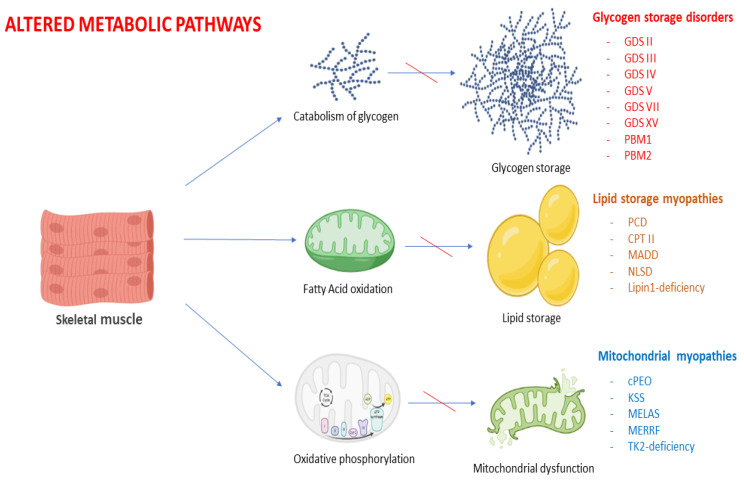
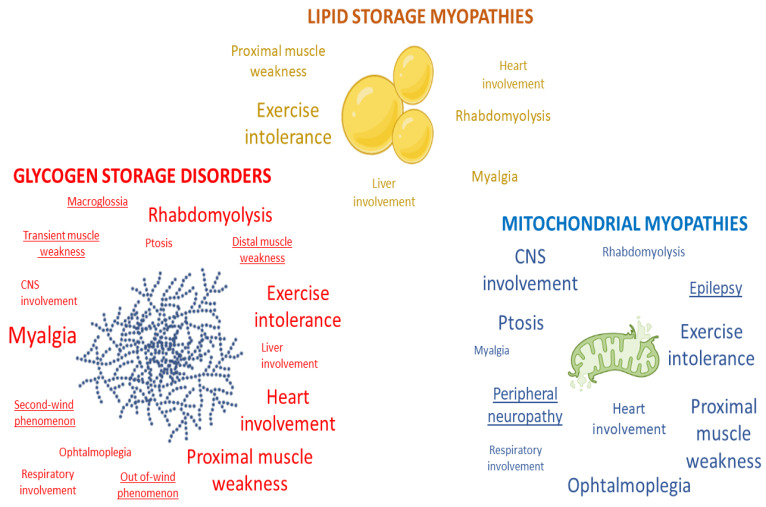
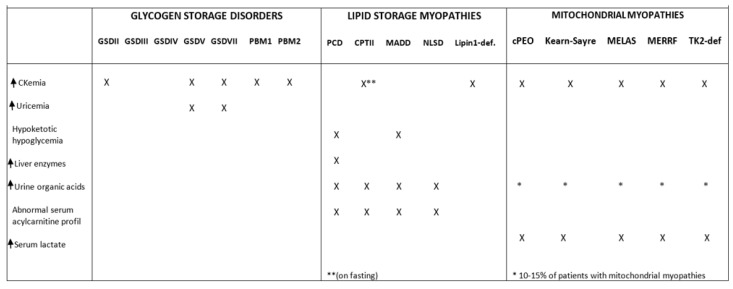
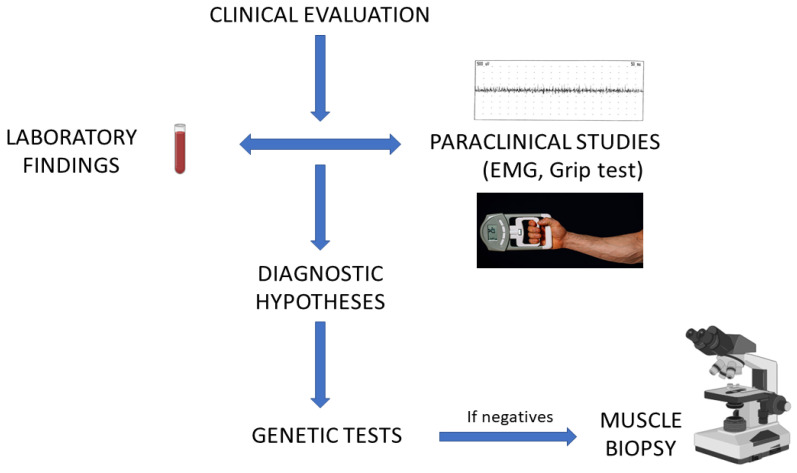
References
-
- Parikh S., Goldstein A., Koenig M.K., Scaglia F., Enns G.M., Saneto R., Anselm I., Cohen B.H., Falk M.J., Greene C., et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Anesthesia Analg. 2014;17:689–701. doi: 10.1038/gim.2014.177. - DOI - PMC - PubMed
-
- Byrne B., Bratkovic D., Díaz-Manera J., Laforêt P., Mozaffar T., van der Ploeg A., Roberts M., Schoser B., Toscano A., Jiang H., et al. Cipaglucosidase alfa/miglustat versus alglucosidase alfa/placebo in late-onset Pompe disease (LOPD): PROPEL study subgroup analyses. Mol. Genet. Metab. 2022;135:S27–S28. doi: 10.1016/j.ymgme.2021.11.054. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
