

Genetic and Clinical Profile of Retinopathies Due to Disease-Causing Variants in Leber Congenital Amaurosis (LCA)-Associated Genes in a Large German Cohort
- PMID: 37240262
- PMCID: PMC10219005
- DOI: 10.3390/ijms24108915
Genetic and Clinical Profile of Retinopathies Due to Disease-Causing Variants in Leber Congenital Amaurosis (LCA)-Associated Genes in a Large German Cohort
Abstract
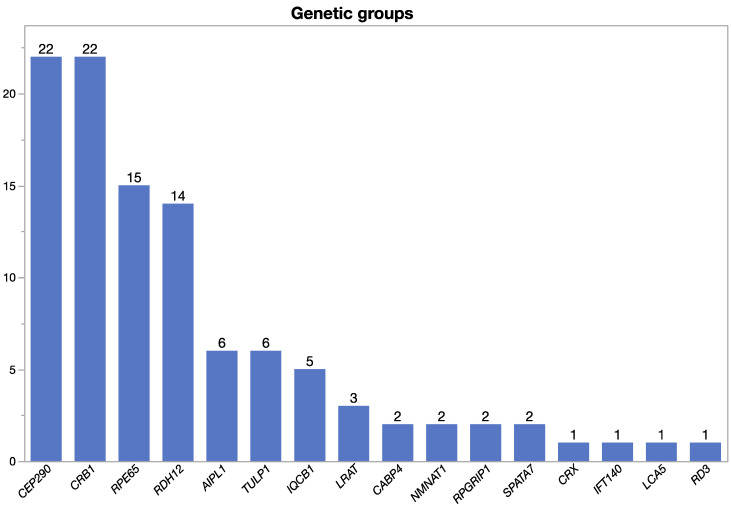
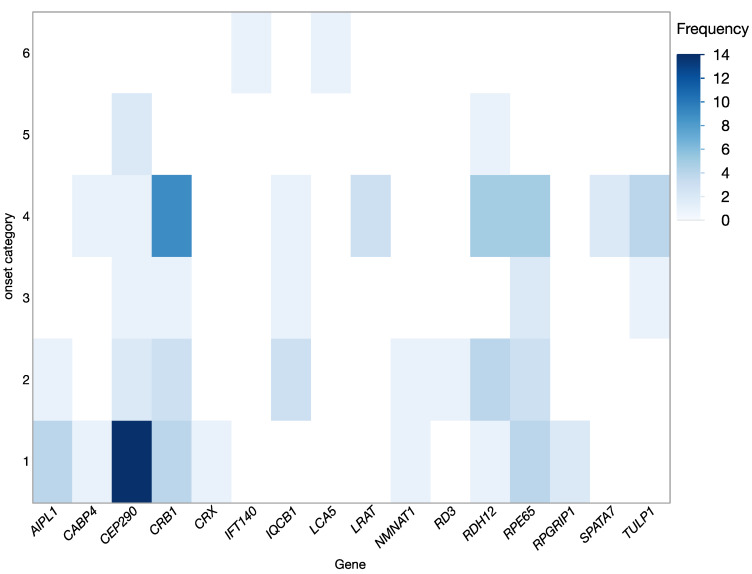
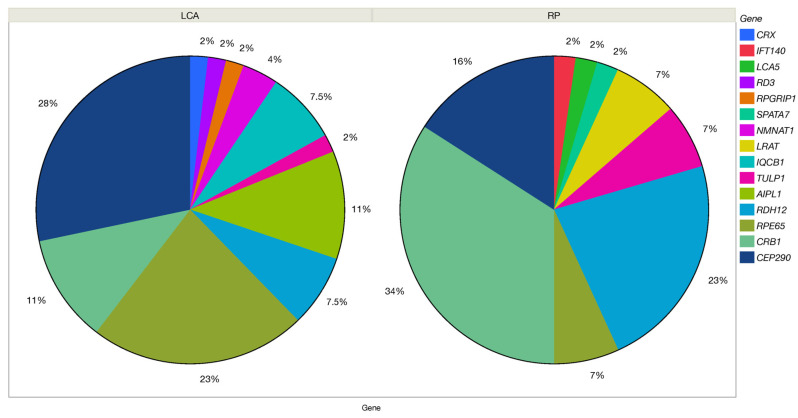
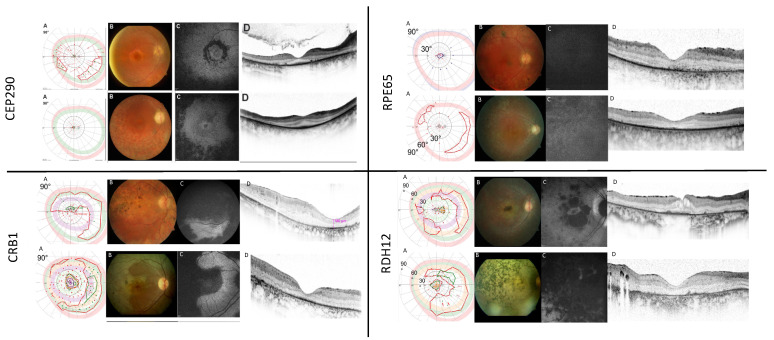
To report the spectrum of Leber congenital amaurosis (LCA) associated genes in a large German cohort and to delineate their associated phenotype. Local databases were screened for patients with a clinical diagnosis of LCA and for patients with disease-causing variants in known LCA-associated genes independent of their clinical diagnosis. Patients with a mere clinical diagnosis were invited for genetic testing. Genomic DNA was either analyzed in a diagnostic-genetic or research setup using various capture panels for syndromic and non-syndromic IRD (inherited retinal dystrophy) genes. Clinical data was obtained mainly retrospectively. Patients with genetic and phenotypic information were eventually included. Descriptive statistical data analysis was performed. A total of 105 patients (53 female, 52 male, age 3-76 years at the time of data collection) with disease-causing variants in 16 LCA-associated genes were included. The genetic spectrum displayed variants in the following genes: CEP290 (21%), CRB1 (21%), RPE65 (14%), RDH12 (13%), AIPL1 (6%), TULP1 (6%), and IQCB1 (5%), and few cases harbored pathogenic variants in LRAT, CABP4, NMNAT1, RPGRIP1, SPATA7, CRX, IFT140, LCA5, and RD3 (altogether accounting for 14%). The most common clinical diagnosis was LCA (53%, 56/105) followed by retinitis pigmentosa (RP, 40%, 42/105), but also other IRDs were seen (cone-rod dystrophy, 5%; congenital stationary night blindness, 2%). Among LCA patients, 50% were caused by variants in CEP290 (29%) and RPE65 (21%), whereas variants in other genes were much less frequent (CRB1 11%, AIPL1 11%, IQCB1 9%, and RDH12 7%, and sporadically LRAT, NMNAT1, CRX, RD3, and RPGRIP1). In general, the patients showed a severe phenotype hallmarked by severely reduced visual acuity, concentric narrowing of the visual field, and extinguished electroretinograms. However, there were also exceptional cases with best corrected visual acuity as high as 0.8 (Snellen), well-preserved visual fields, and preserved photoreceptors in spectral domain optical coherence tomography. Phenotypic variability was seen between and within genetic subgroups. The study we are presenting pertains to a considerable LCA group, furnishing valuable comprehension of the genetic and phenotypic spectrum. This knowledge holds significance for impending gene therapeutic trials. In this German cohort, CEP290 and CRB1 are the most frequently mutated genes. However, LCA is genetically highly heterogeneous and exhibits clinical variability, showing overlap with other IRDs. For any therapeutic gene intervention, the disease-causing genotype is the primary criterion for treatment access, but the clinical diagnosis, state of the retina, number of to be treated target cells, and the time point of treatment will be crucial.
Keywords: LCA-associated genes; Leber congenital amaurosis (LCA); genotype-phenotype correlation; nherited retinal dystrophies; retinitis pigmentosa (RP).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Leber T.G. Ueber Retinitis pigmentosa und angeborene Amaurose. Graefes Archive of Clincal Experimental Ophthalmology. Arch. Für Ophthalmol. 1869;15:1–25.
-
- Retina P. 2023. [(accessed on 1 March 2023)]. Available online: https://www.pro-retina.de.
-
- Sheck L., Davies W.I.L., Moradi P., Robson A.G., Kumaran N., Liasis A.C., Webster A.R., Moore A.T., Michaelides M. Leber Congenital Amaurosis Associated with Mutations in CEP290, Clinical Phenotype, and Natural History in Preparation for Trials of Novel Therapies. Ophthalmology. 2018;125:894–903. doi: 10.1016/j.ophtha.2017.12.013. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
