

Single-cell transcriptomics reveals immune suppression and cell states predictive of patient outcomes in rhabdomyosarcoma
- PMID: 37244912
- PMCID: PMC10224926
- DOI: 10.1038/s41467-023-38886-8
Single-cell transcriptomics reveals immune suppression and cell states predictive of patient outcomes in rhabdomyosarcoma
Abstract
Paediatric rhabdomyosarcoma (RMS) is a soft tissue malignancy of mesenchymal origin that is thought to arise as a consequence of derailed myogenic differentiation. Despite intensive treatment regimens, the prognosis for high-risk patients remains dismal. The cellular differentiation states underlying RMS and how these relate to patient outcomes remain largely elusive. Here, we use single-cell mRNA sequencing to generate a transcriptomic atlas of RMS. Analysis of the RMS tumour niche reveals evidence of an immunosuppressive microenvironment. We also identify a putative interaction between NECTIN3 and TIGIT, specific to the more aggressive fusion-positive (FP) RMS subtype, as a potential cause of tumour-induced T-cell dysfunction. In malignant RMS cells, we define transcriptional programs reflective of normal myogenic differentiation and show that these cellular differentiation states are predictive of patient outcomes in both FP RMS and the less aggressive fusion-negative subtype. Our study reveals the potential of therapies targeting the immune microenvironment of RMS and suggests that assessing tumour differentiation states may enable a more refined risk stratification.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
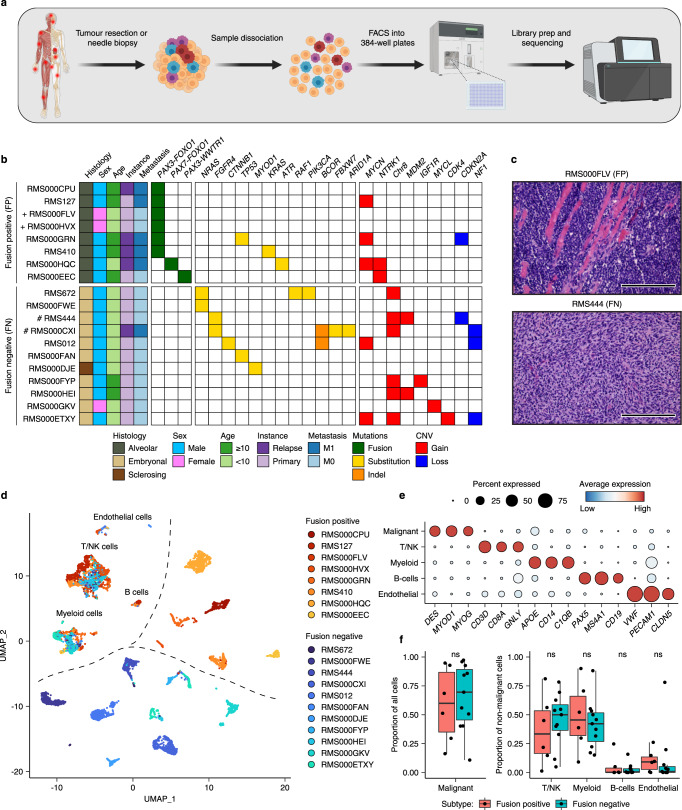
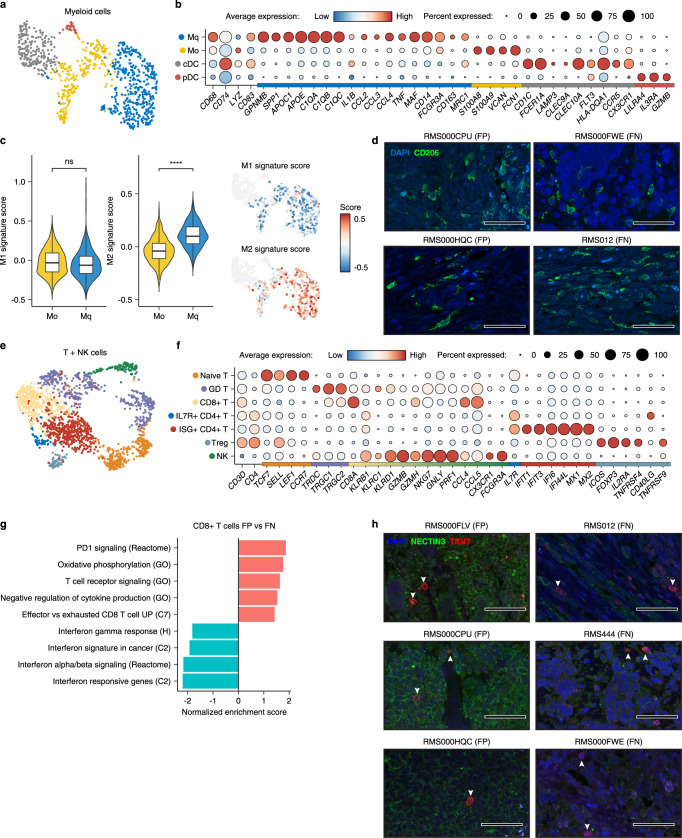

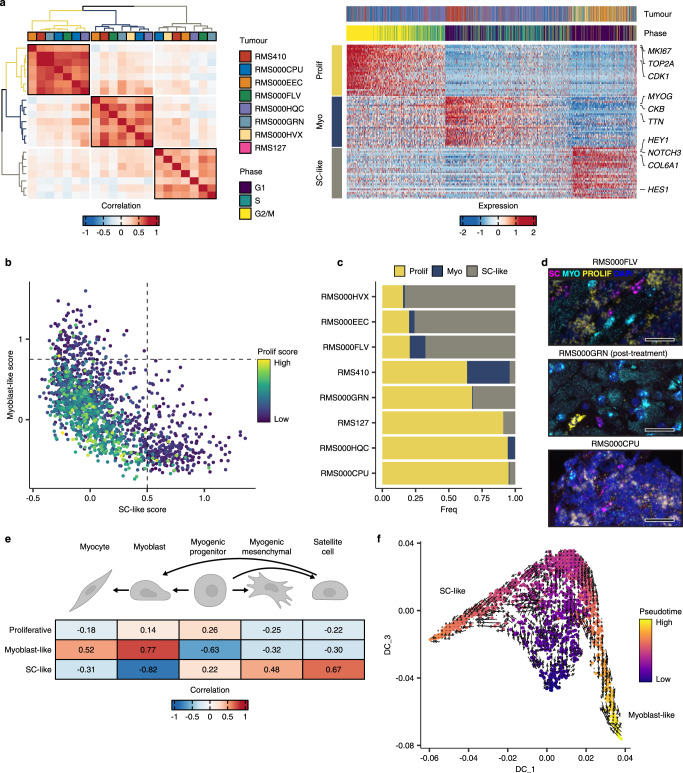
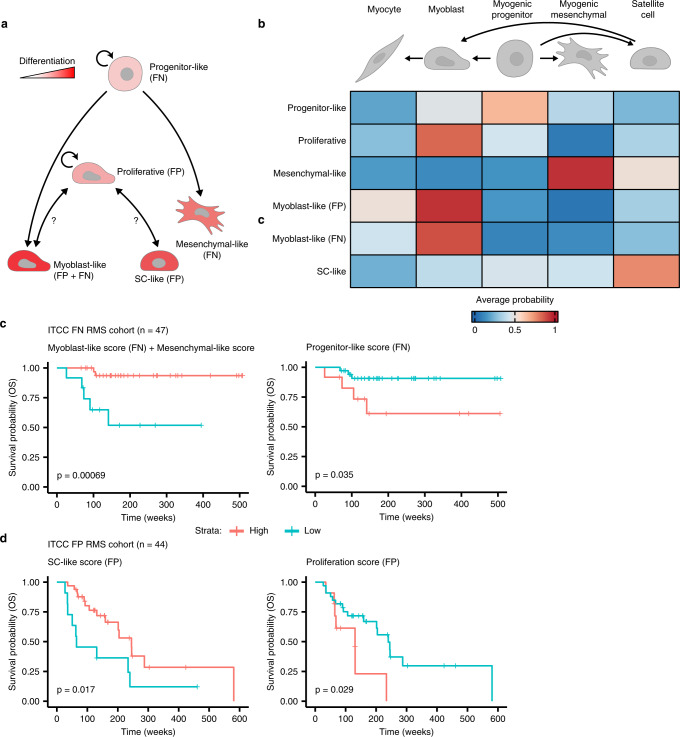
References
-
- Stiller D, Holzhausen HJ. Ultrastructural organization of cell characteristics relevant to diagnosis in rhabdomyosarcomas. Zentralblatt f.ür. Allg. Pathologie Pathologische Anat. 1988;134:449–466. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
