

Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance
- PMID: 37248301
- PMCID: PMC11421770
- DOI: 10.1038/s41591-023-02371-y
Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance
Abstract
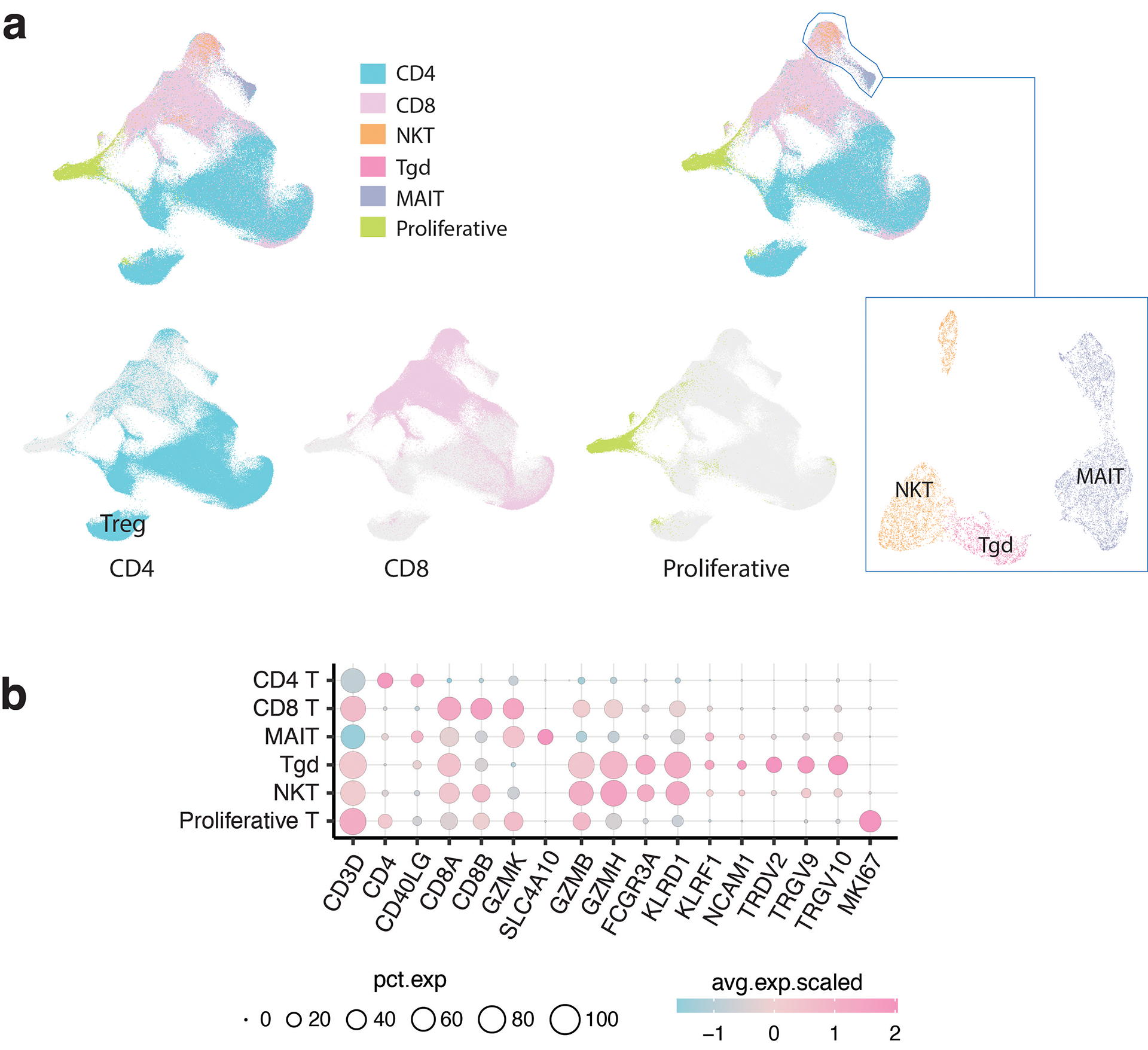
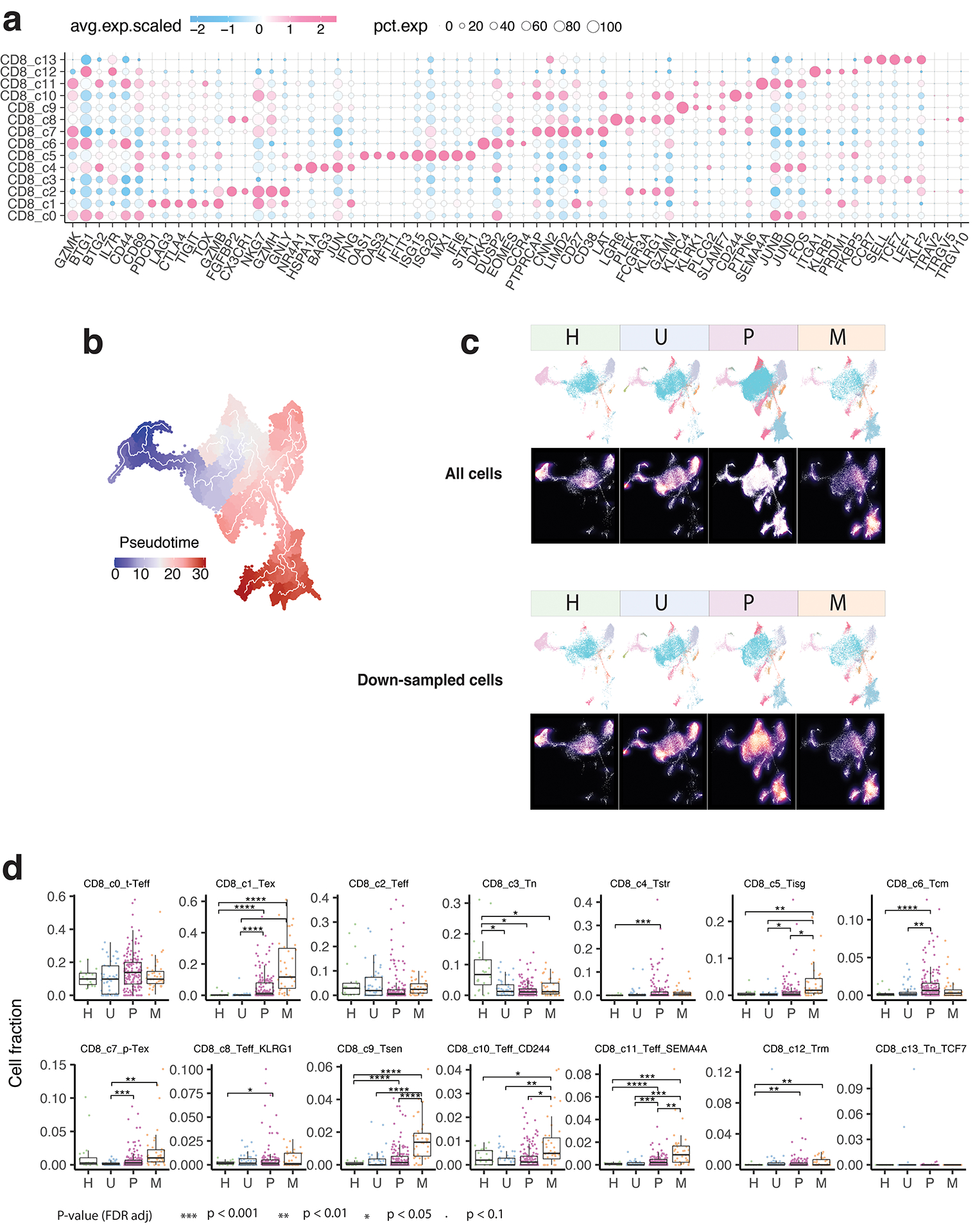
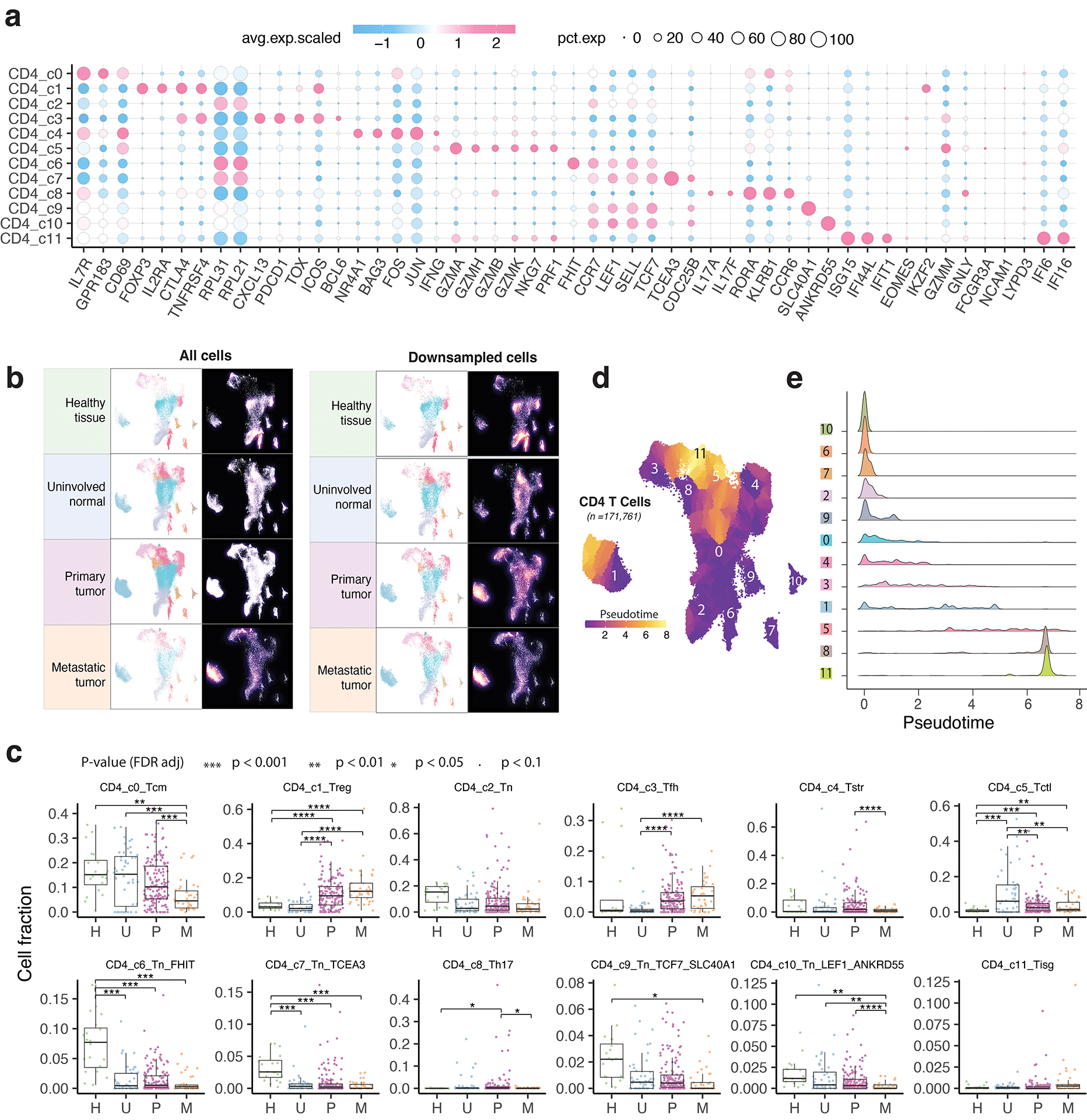
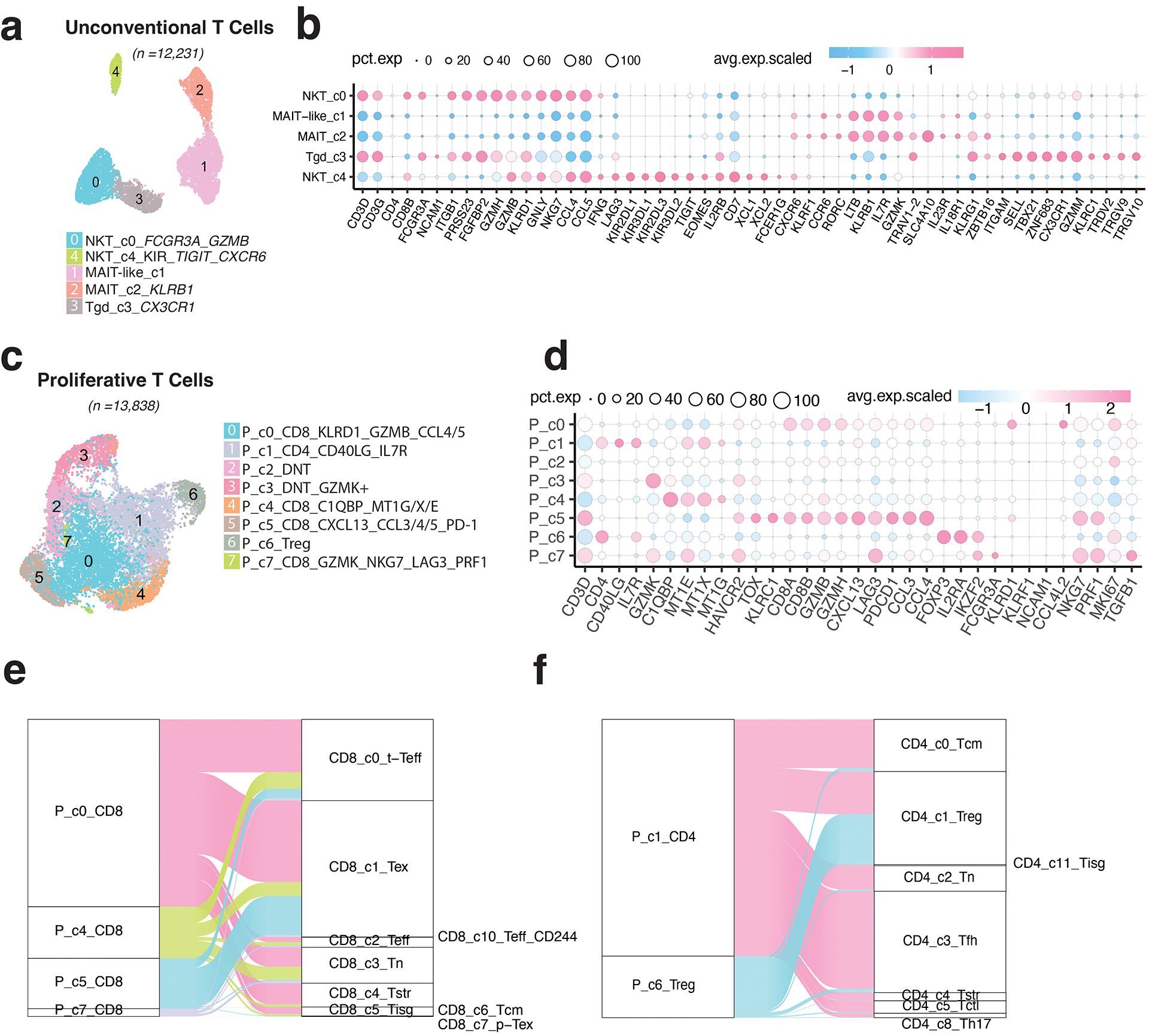
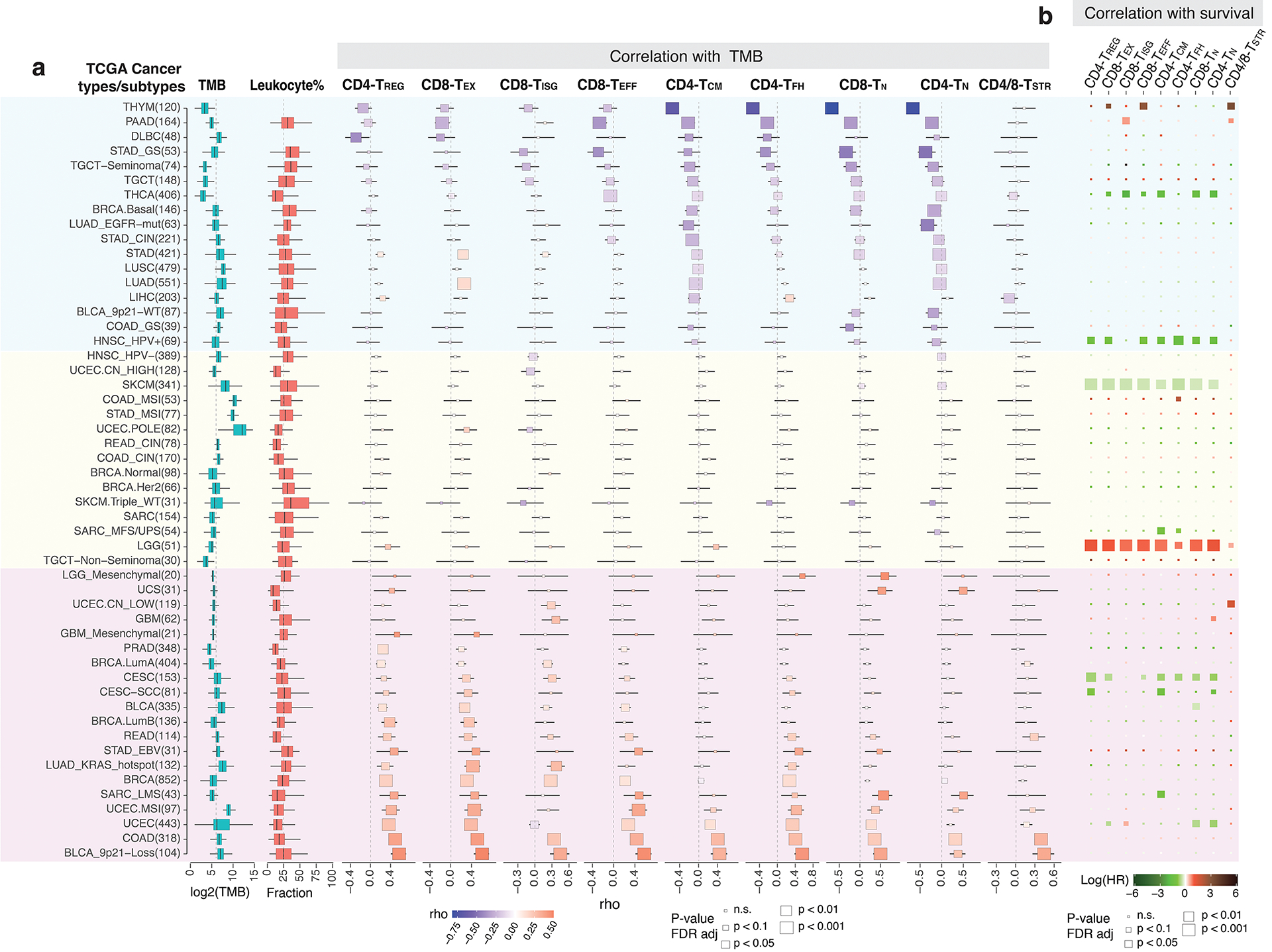
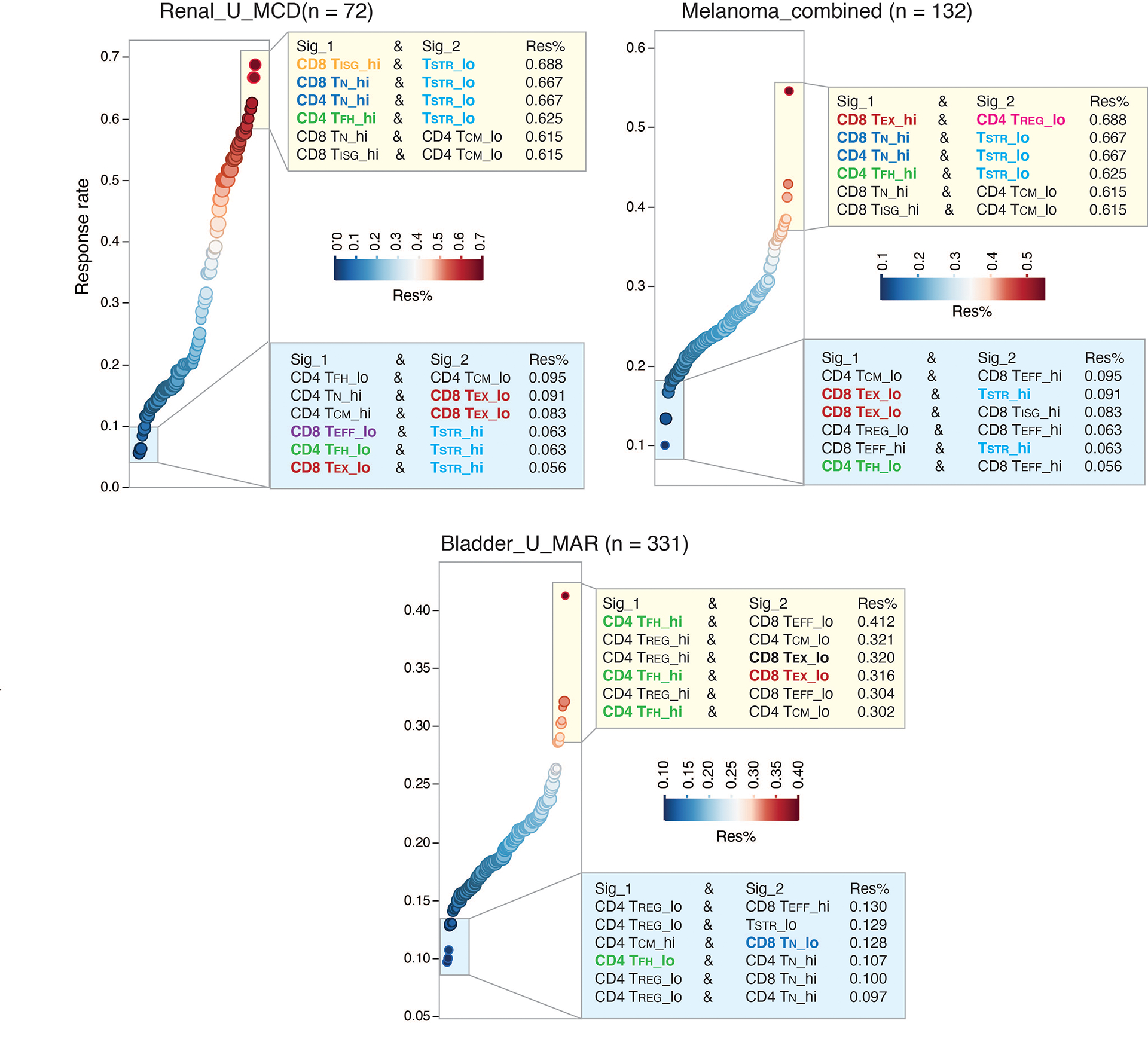
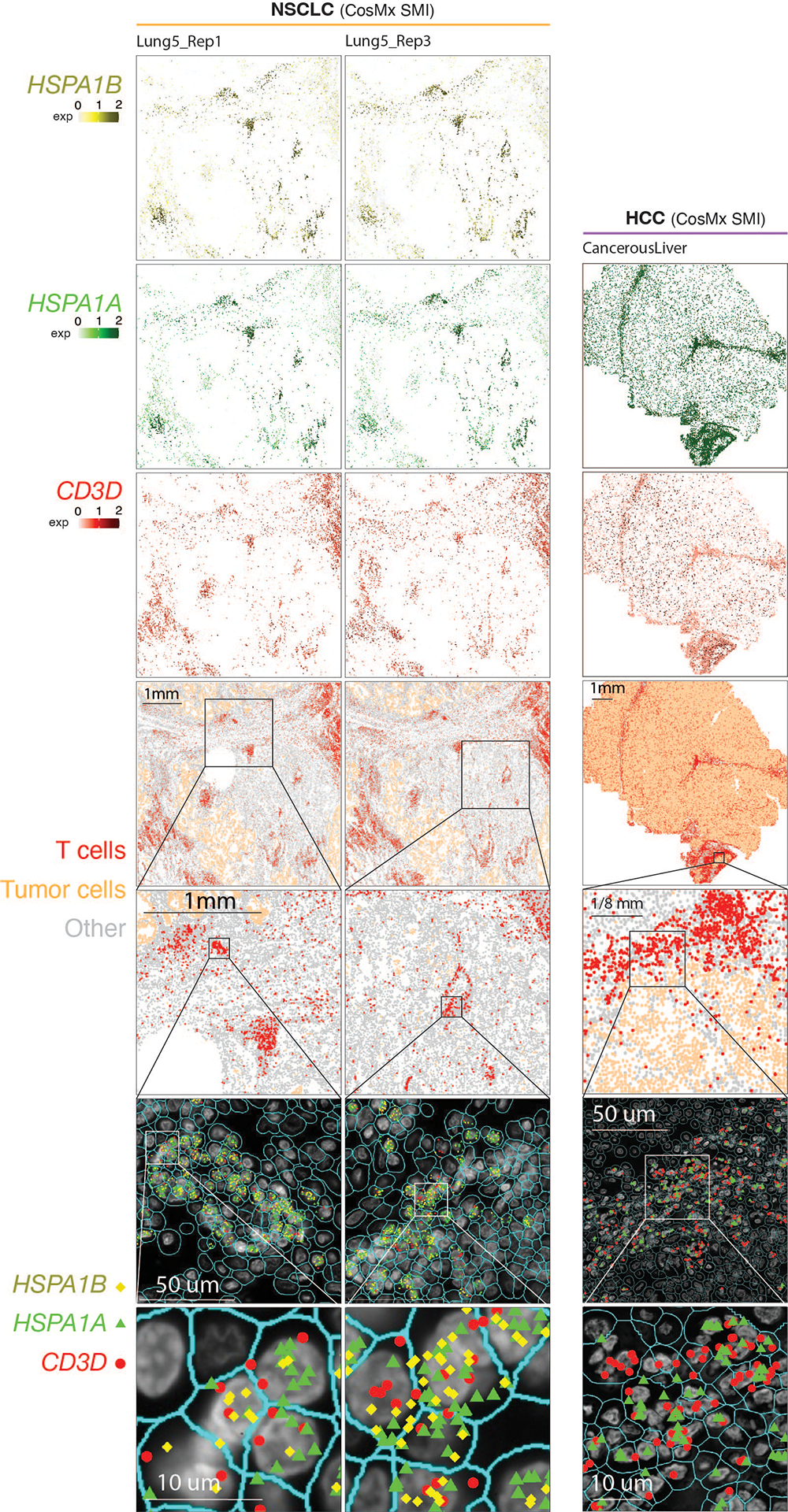
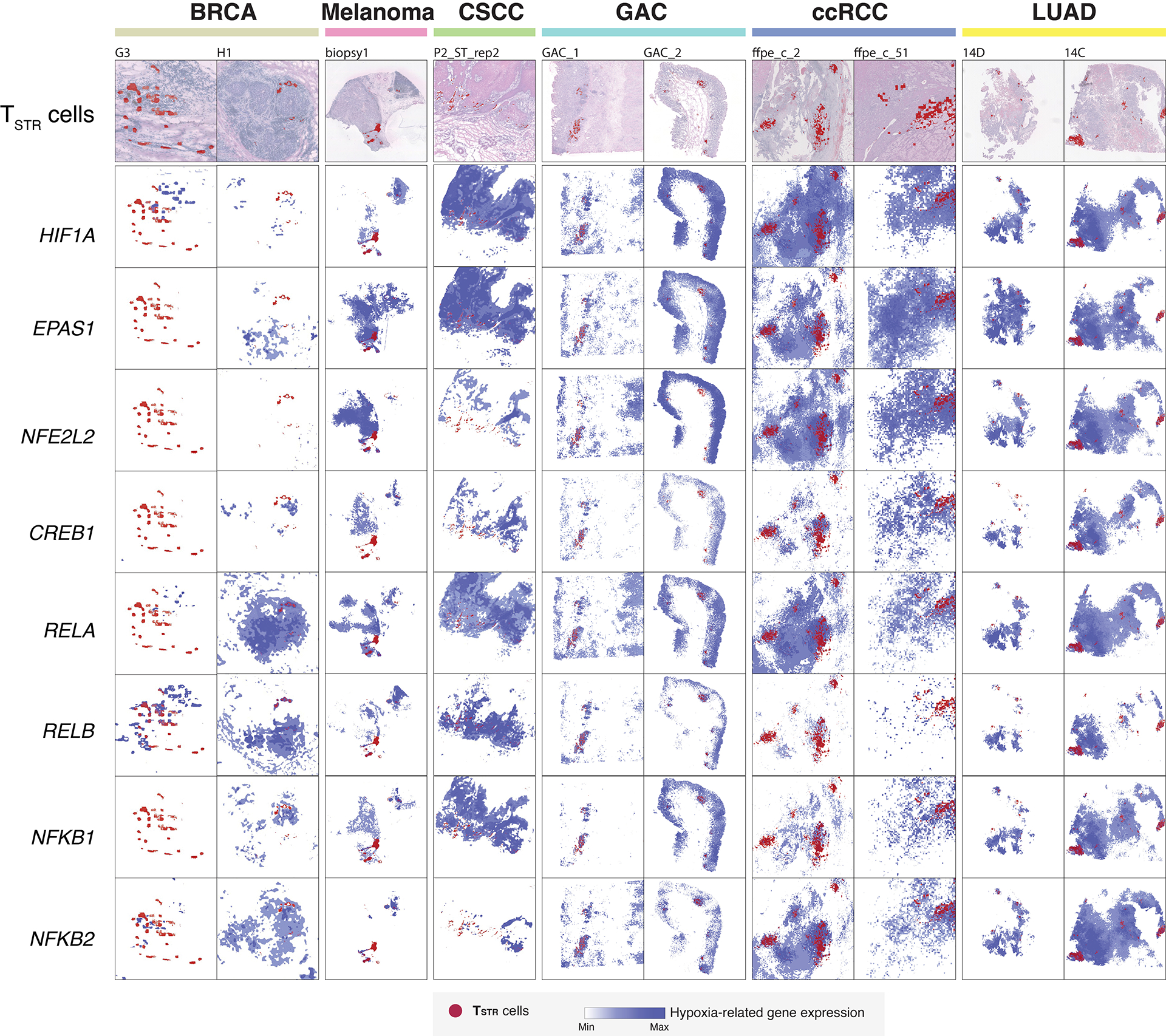
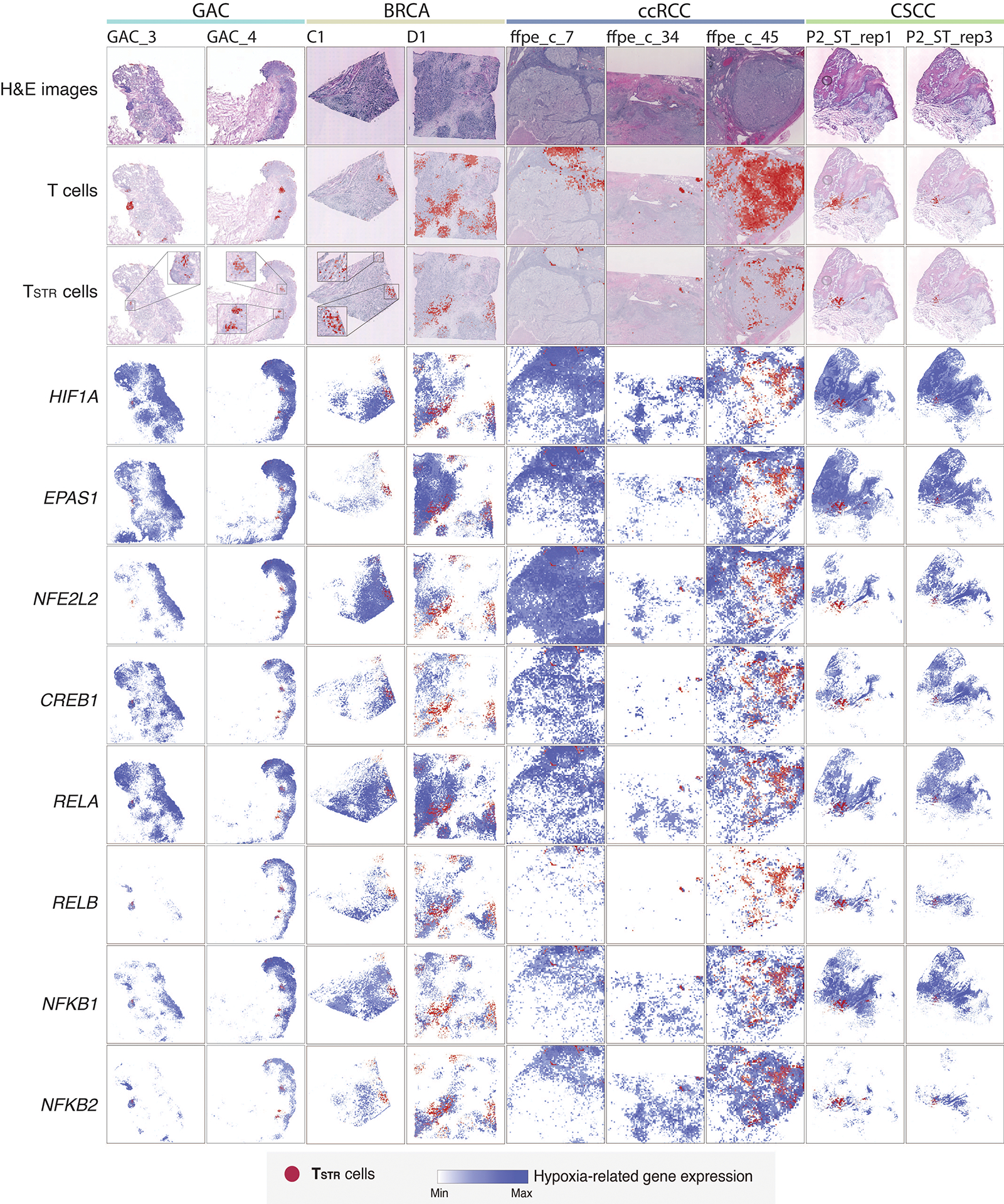
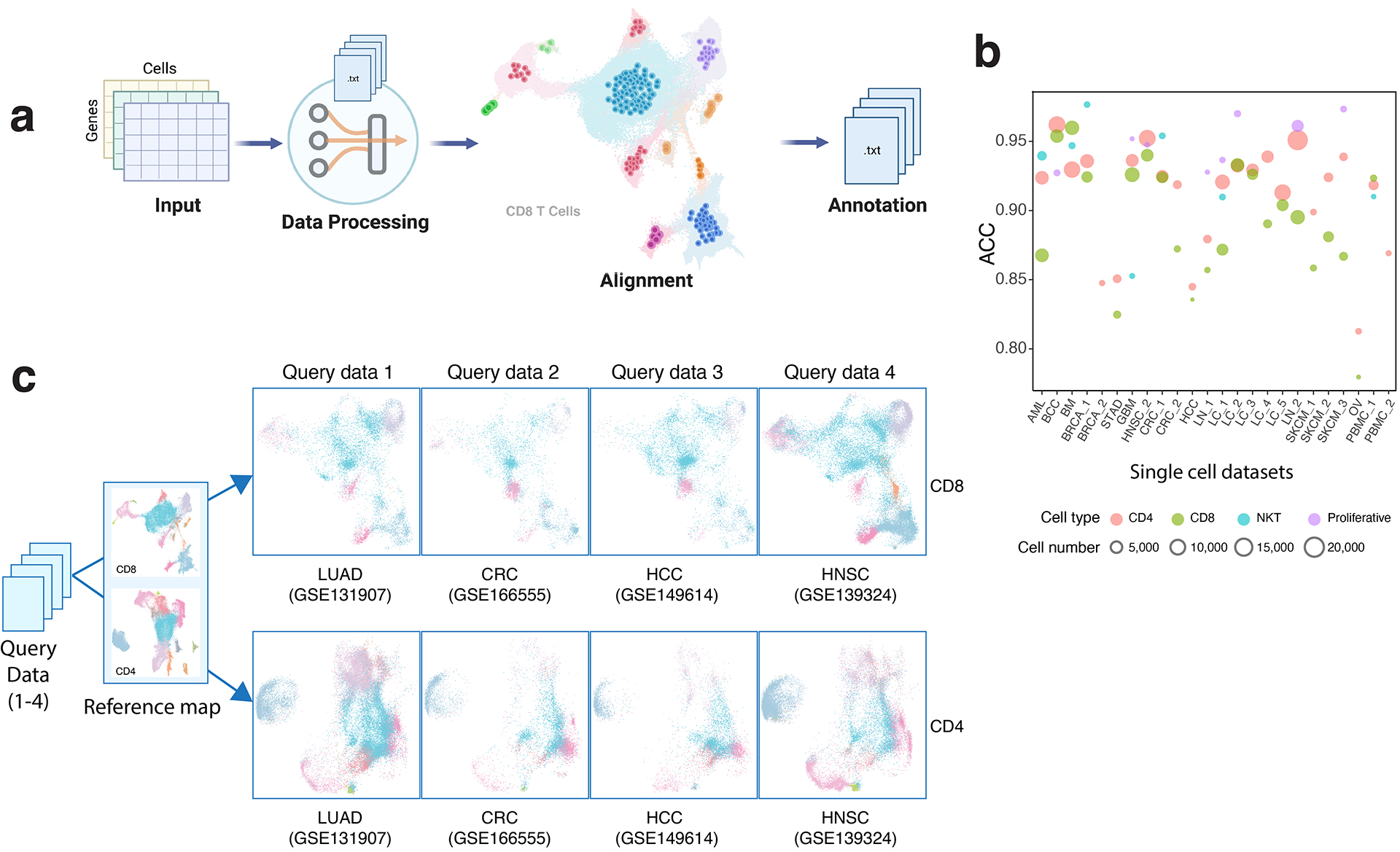
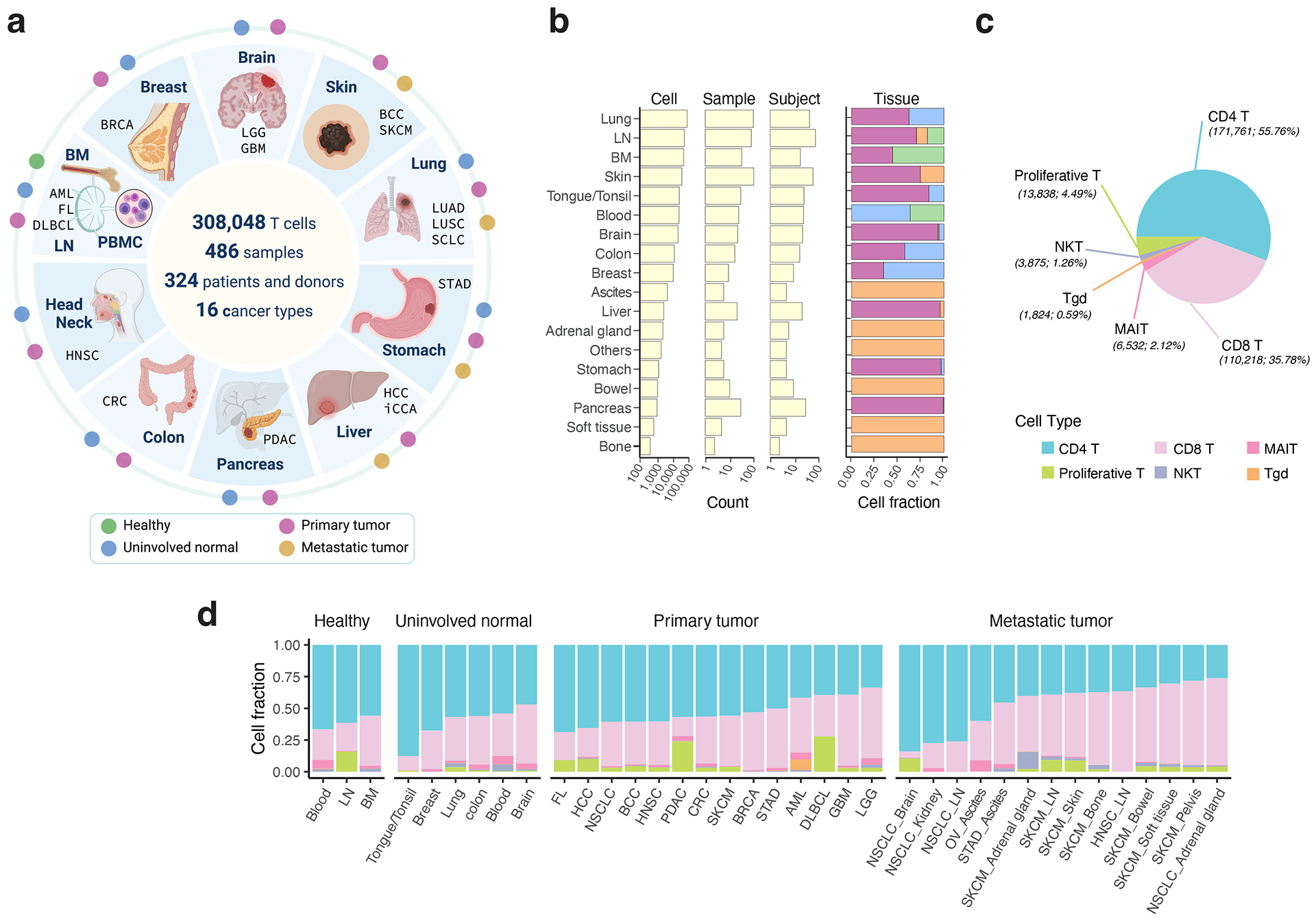
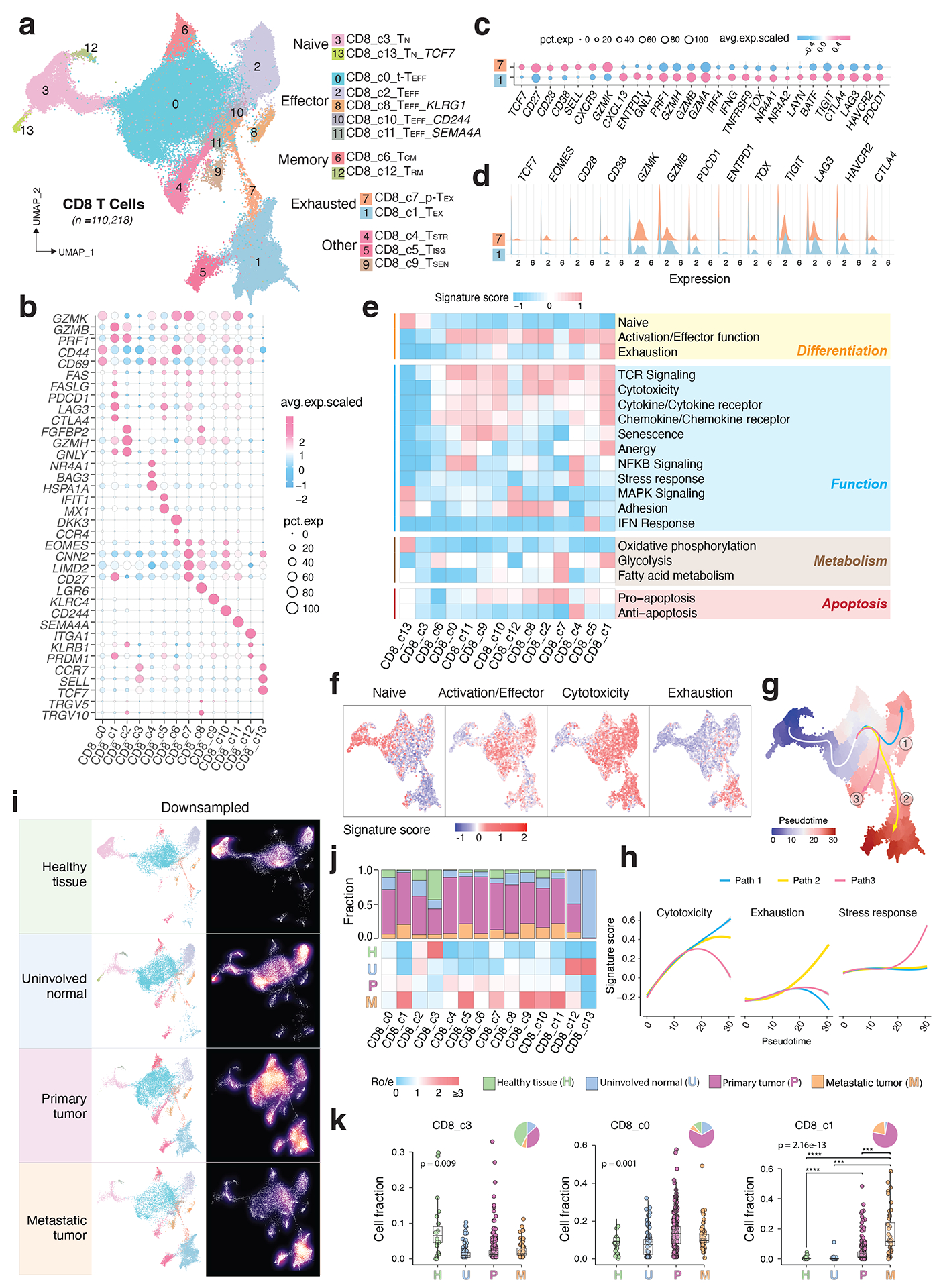
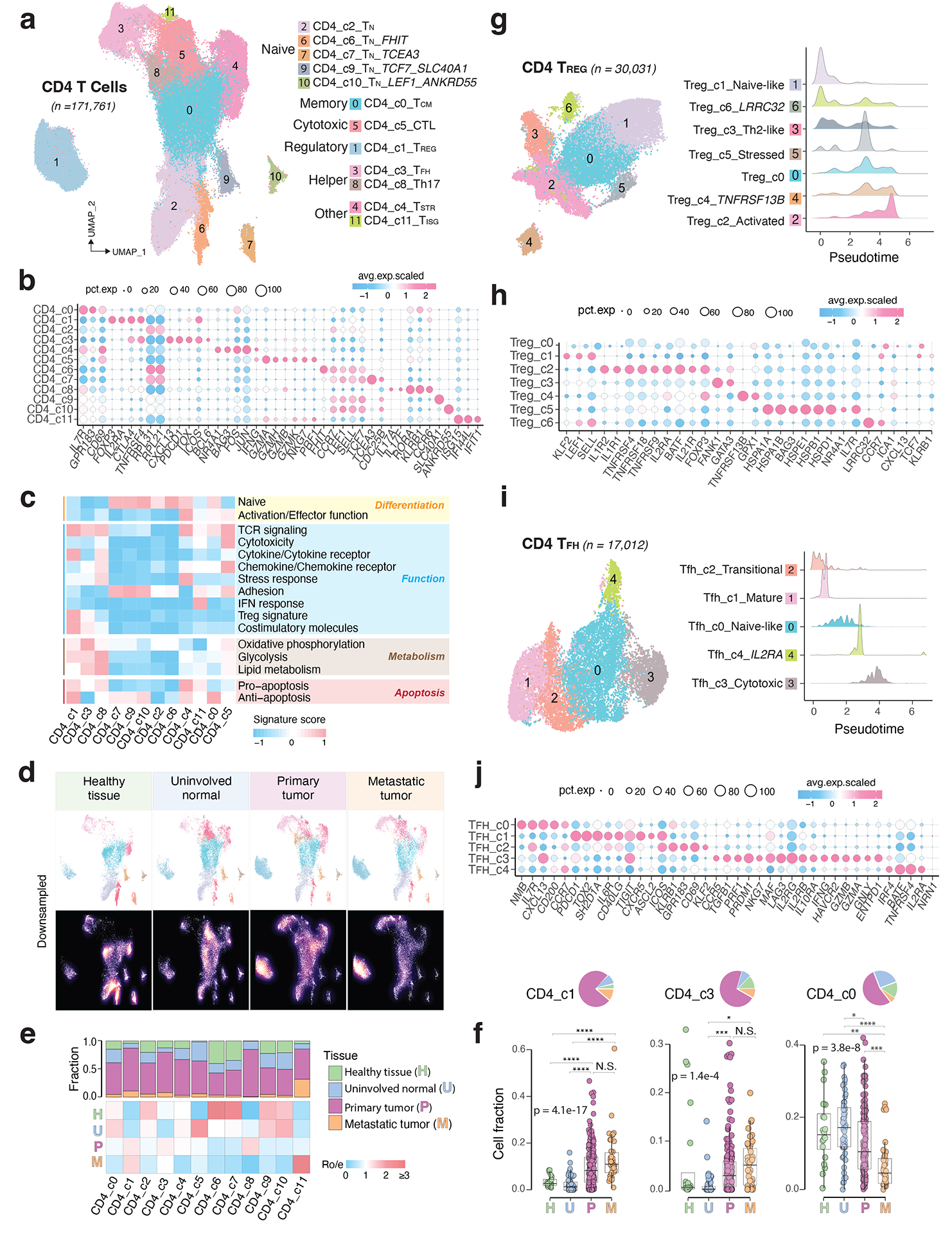
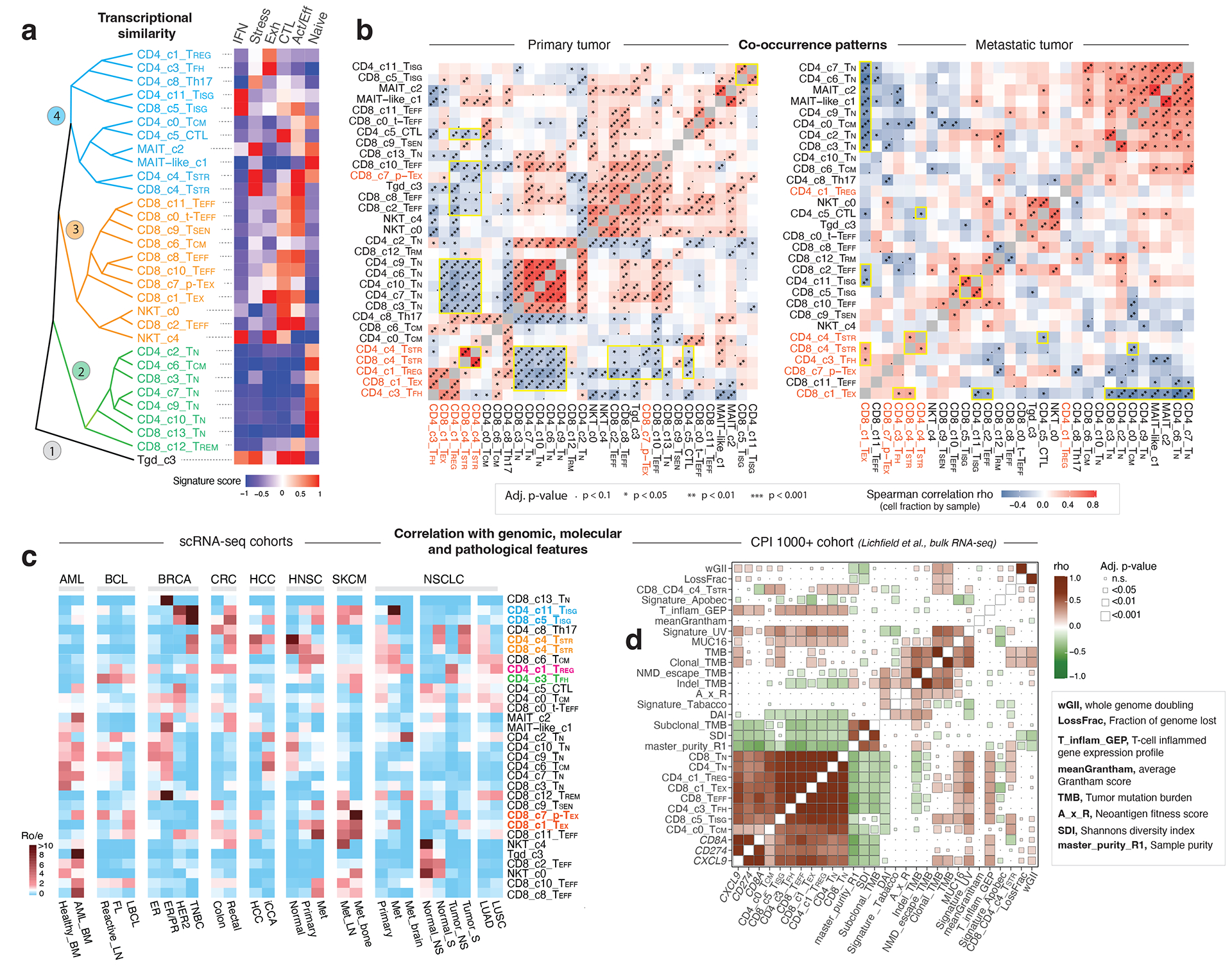
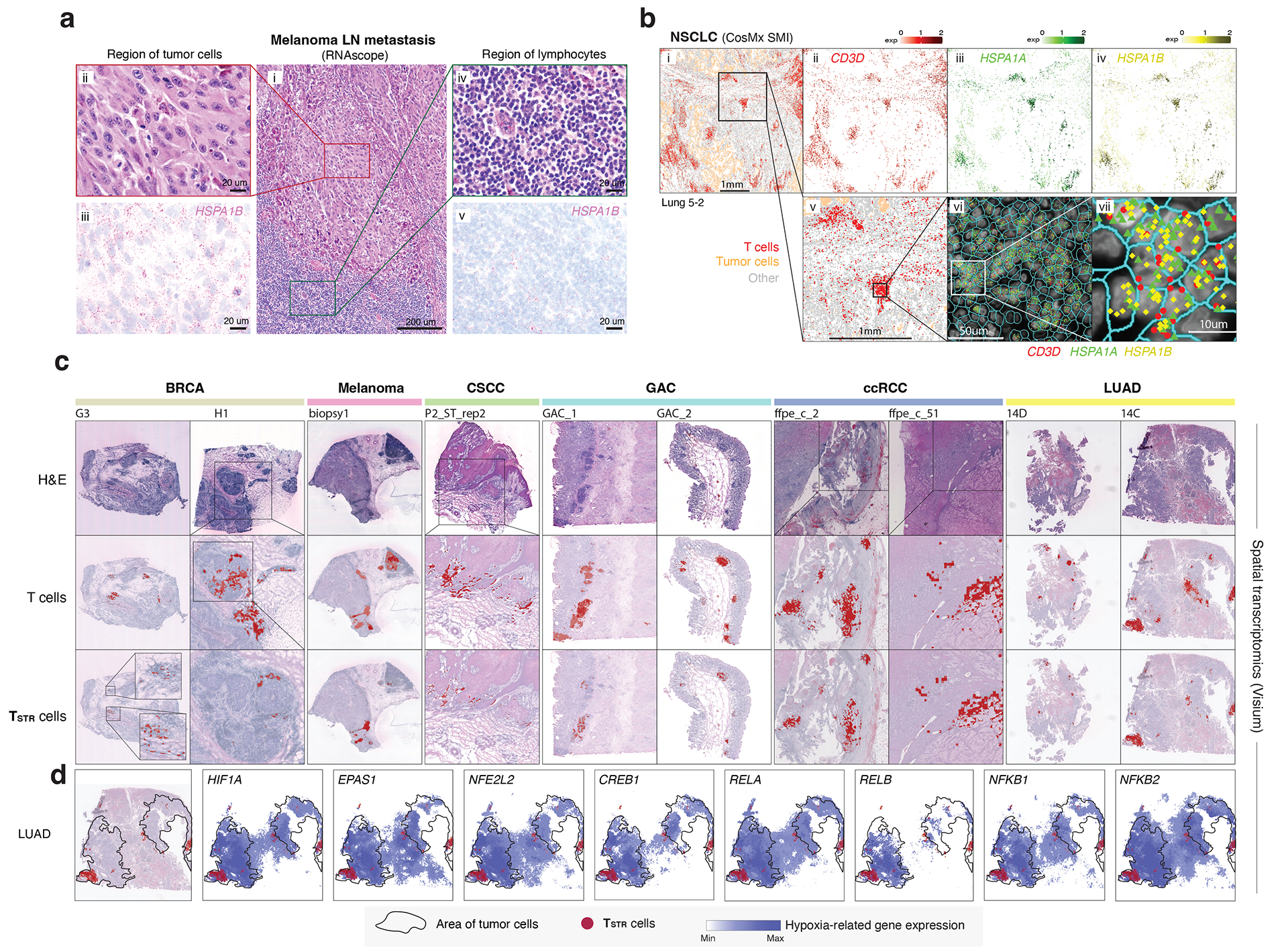
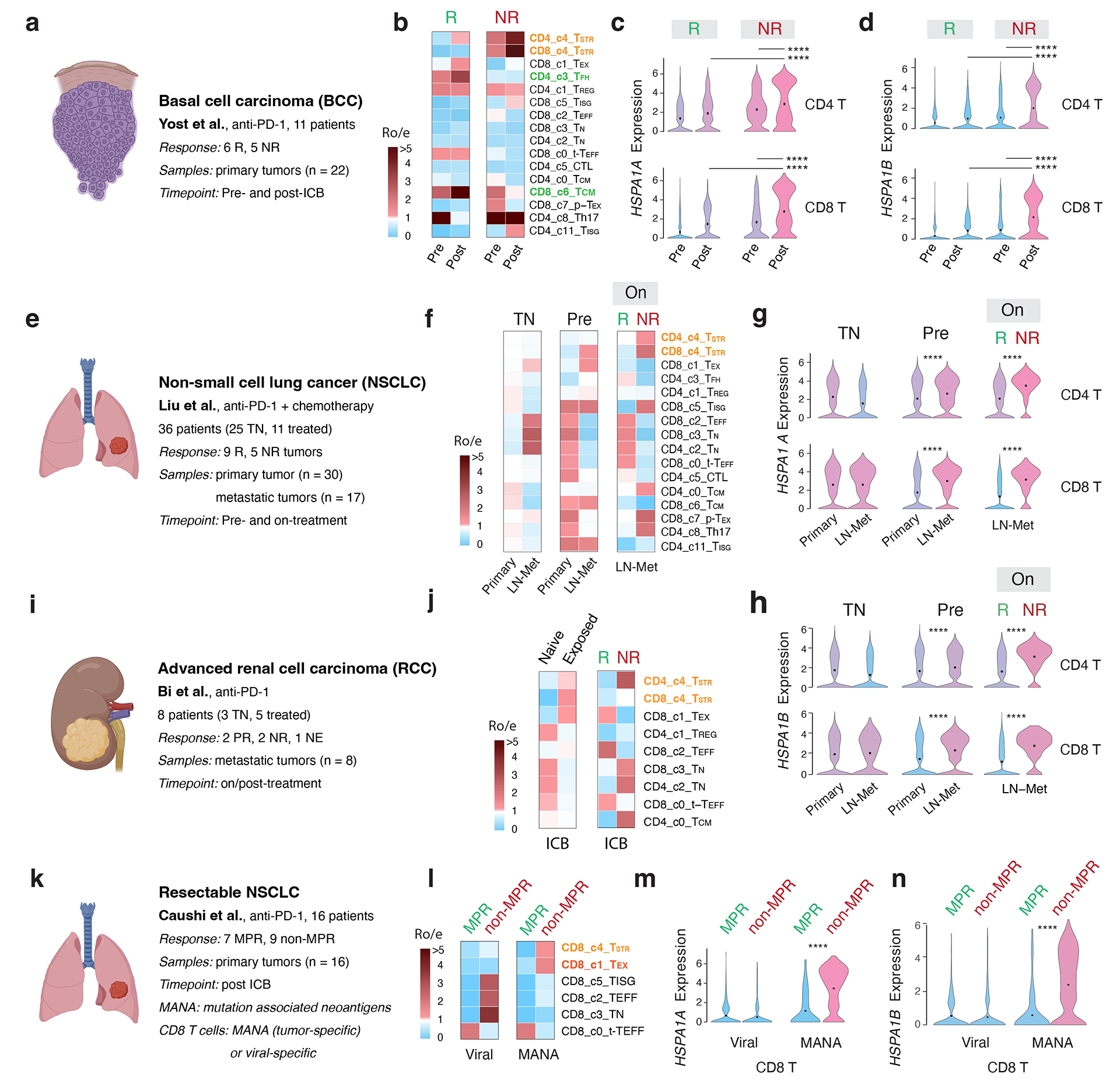
Tumor-infiltrating T cells offer a promising avenue for cancer treatment, yet their states remain to be fully characterized. Here we present a single-cell atlas of T cells from 308,048 transcriptomes across 16 cancer types, uncovering previously undescribed T cell states and heterogeneous subpopulations of follicular helper, regulatory and proliferative T cells. We identified a unique stress response state, TSTR, characterized by heat shock gene expression. TSTR cells are detectable in situ in the tumor microenvironment across various cancer types, mostly within lymphocyte aggregates or potential tertiary lymphoid structures in tumor beds or surrounding tumor edges. T cell states/compositions correlated with genomic, pathological and clinical features in 375 patients from 23 cohorts, including 171 patients who received immune checkpoint blockade therapy. We also found significantly upregulated heat shock gene expression in intratumoral CD4/CD8+ cells following immune checkpoint blockade treatment, particularly in nonresponsive tumors, suggesting a potential role of TSTR cells in immunotherapy resistance. Our well-annotated T cell reference maps, web portal and automatic alignment/annotation tool could provide valuable resources for T cell therapy optimization and biomarker discovery.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
-
- Russell JH & Ley TJ Lymphocyte-mediated cytotoxicity. Annual review of immunology 20, 323–370 (2002). - PubMed
-
- Fridman WH, Pagès F, Sautès-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nature Reviews Cancer 12, 298–306 (2012). - PubMed
-
- Janssen EM, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 434, 88–93 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
