

Hypophosphatasia: from birth to adulthood
- PMID: 37249457
- PMCID: PMC10665056
- DOI: 10.20945/2359-3997000000626
Hypophosphatasia: from birth to adulthood
Abstract
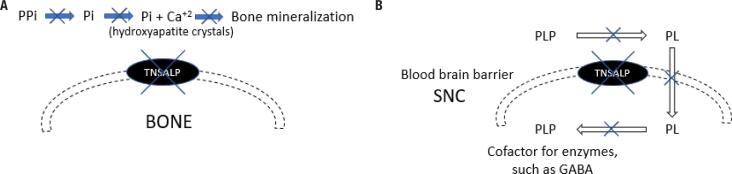
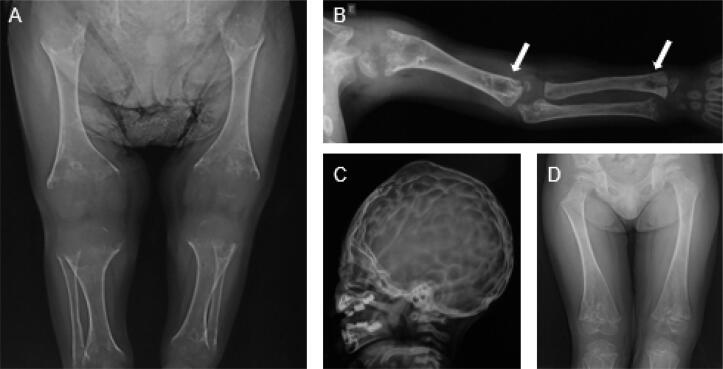
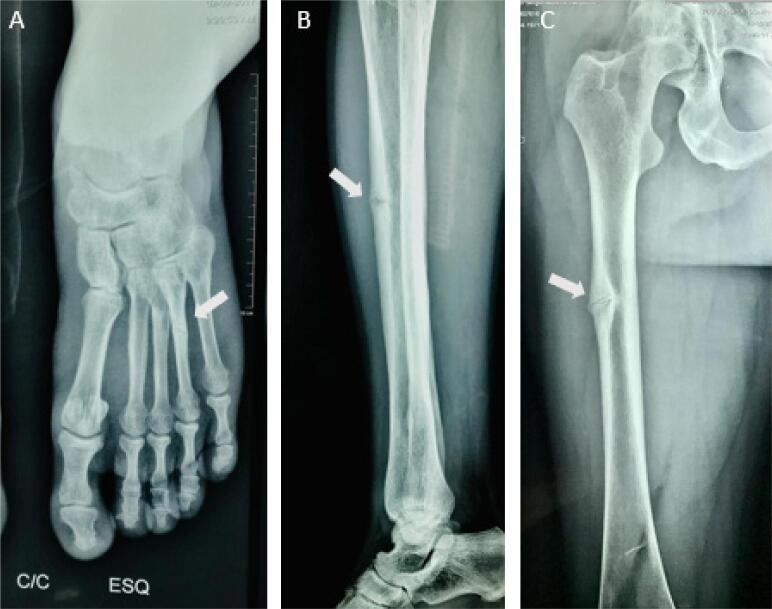
Hypophosphatasia (HPP) is an inherited disease caused by a low activity of tissue-nonspecific alkaline phosphatase, a hydrolase that removes phosphate groups from many molecules. Decreased alkaline phosphatase activity leads to the accumulation of three main metabolites, i.e., pyridoxal 5´-phosphate (PLP), inorganic pyrophosphate (PPi), and phosphoethanolamine. Impairment in PLP dephosphorylation induces seizures, while PPi accumulation inhibits bone mineralization. Clinically, HPP has a wide spectrum of presentations, ranging from neonatal death to an apparent lack of symptoms. This disease is classified into six subtypes according to the age at onset of first signs or symptoms. The clinical manifestations of the disease include rickets-like bone changes, bone demineralization, fragility fractures, reduced muscular strength, chest deformity, pulmonary hypoplasia, nephrolithiasis, nephrocalcinosis, and chondrocalcinosis. Treatment of HPP consists of enzyme replacement therapy. Before this therapy was approved, treatment was palliative and associated with high morbidity and mortality. Asfotase alfa has changed the prognosis of the disease by reducing bone deformity and improving bone mineralization, lung function, and muscle weakness, among other benefits. In adults, teriparatide and anti-sclerostin antibody have been used off-label in selected cases, demonstrating benefit in accelerating fracture healing and in concomitant treatment of osteoporosis. This review summarizes the main aspects of HPP and identifies the particularities of the disease in adult patients.
Keywords: Hypophosphatasia; TNSALP mutation; alkaline phosphatase; asfotase alfa.
Conflict of interest statement
Disclosure: MLC has received fees from Alexion Pharmaceuticals for consulting and speaking. FRS has no potential conflict of interest relevant to this article.
Figures
References
-
- Rathbun JC. Hypophosphatasia: a new developmental anomaly. Am J Dis Child. 1948;75:822–831. - PubMed
-
- Buchet R, Millán JL, Magne D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol. 2013;1053:27–51. - PubMed
-
- Weiss MJ, Ray K, Henthorn PS, Lamb B, Kadesch T, Harris H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J Biol Chem. 1988;263(24):12002–12010. - PubMed
-
- Whyte MP, Walkenhorst DA, Fedde KN, Henthorn PS, Hill CS. Hypophosphatasia: levels of bone alkaline phosphatase immunoreactivity in serum reflect disease severity. J Clin Endocrinol Metab. 1996;81(6):2142–2148. - PubMed
-
- Fraser D. Hypophosphatasia. Am J Med. 1957;22(5):730–746. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials