

Rational Chemical Design of Molecular Glue Degraders
- PMID: 37252349
- PMCID: PMC10214506
- DOI: 10.1021/acscentsci.2c01317
Rational Chemical Design of Molecular Glue Degraders
Erratum in
-
Correction to "Rational Chemical Design of Molecular Glue Degraders".ACS Cent Sci. 2023 Jul 21;9(8):1702. doi: 10.1021/acscentsci.3c00844. eCollection 2023 Aug 23. ACS Cent Sci. 2023. PMID: 37637749 Free PMC article.
Abstract
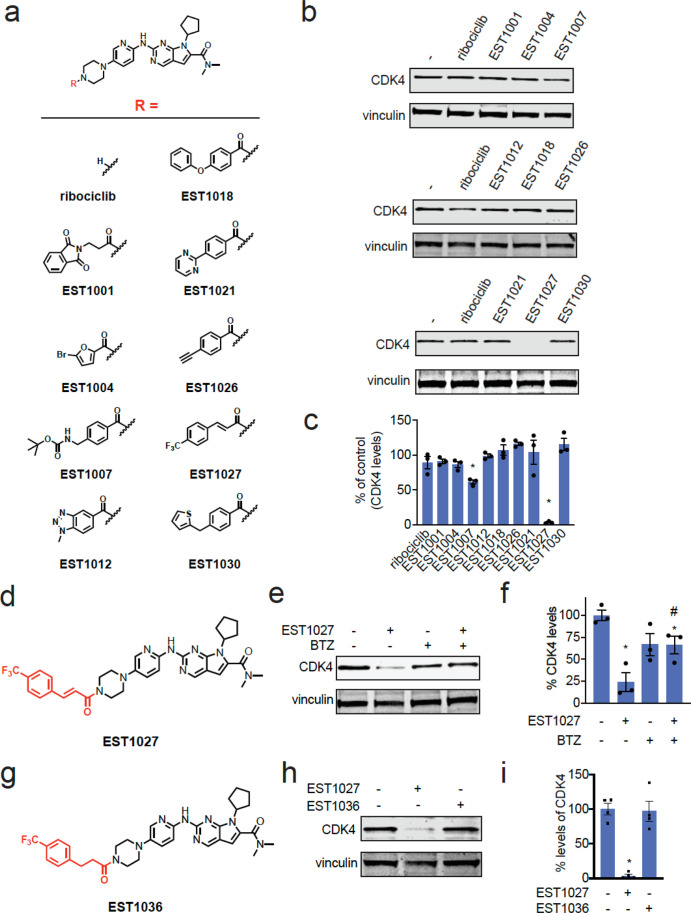
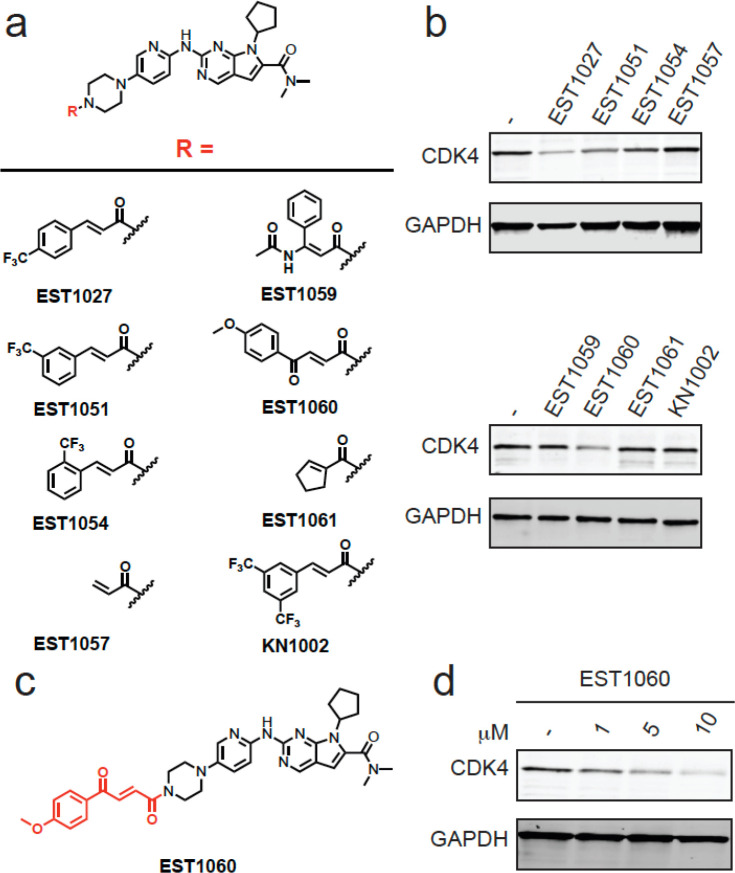
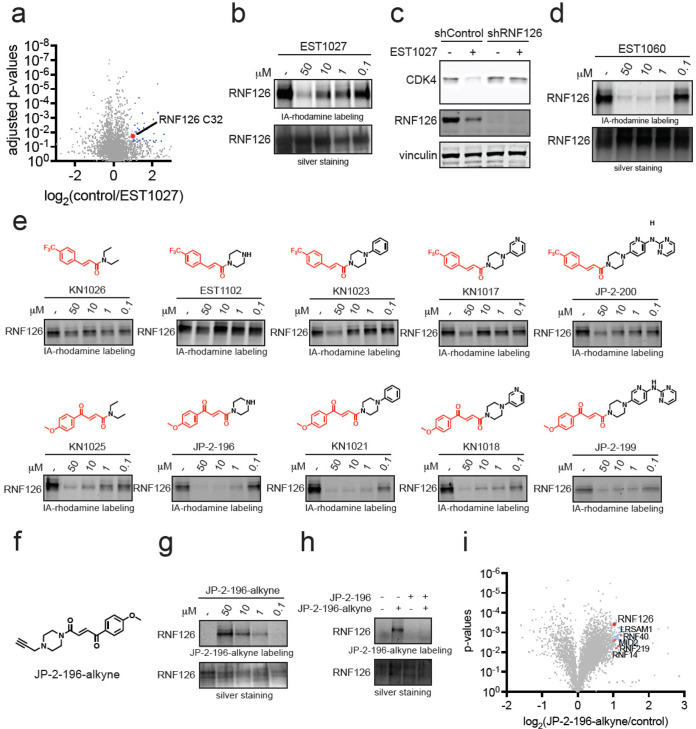
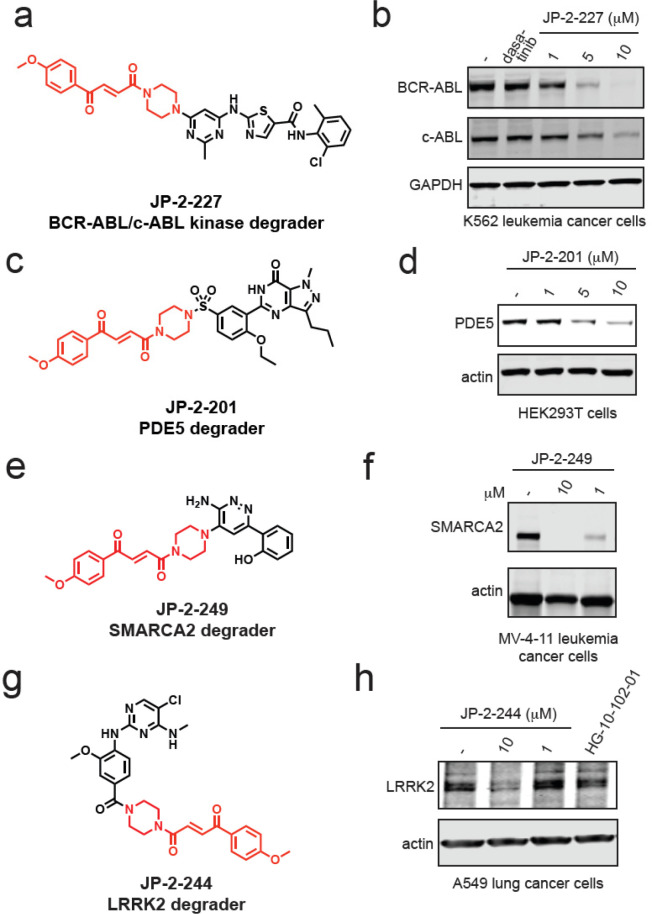
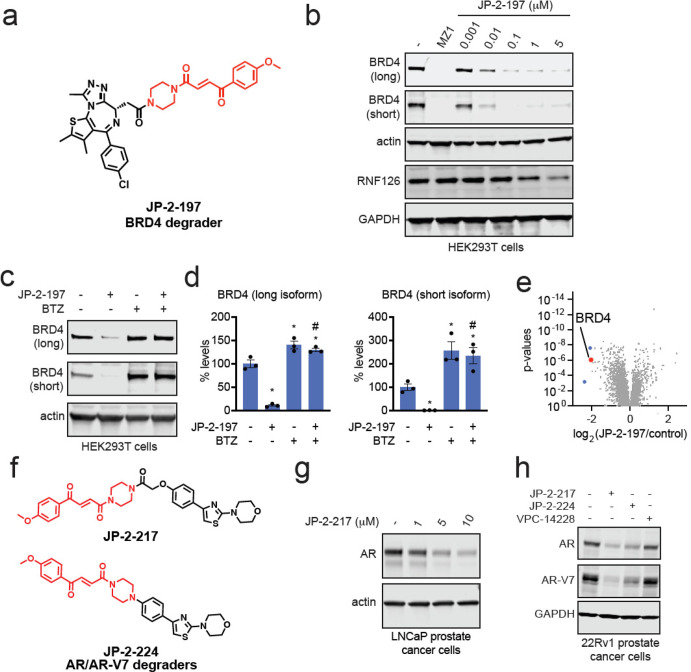
Targeted protein degradation with molecular glue degraders has arisen as a powerful therapeutic modality for eliminating classically undruggable disease-causing proteins through proteasome-mediated degradation. However, we currently lack rational chemical design principles for converting protein-targeting ligands into molecular glue degraders. To overcome this challenge, we sought to identify a transposable chemical handle that would convert protein-targeting ligands into molecular degraders of their corresponding targets. Using the CDK4/6 inhibitor ribociclib as a prototype, we identified a covalent handle that, when appended to the exit vector of ribociclib, induced the proteasome-mediated degradation of CDK4 in cancer cells. Further modification of our initial covalent scaffold led to an improved CDK4 degrader with the development of a but-2-ene-1,4-dione ("fumarate") handle that showed improved interactions with RNF126. Subsequent chemoproteomic profiling revealed interactions of the CDK4 degrader and the optimized fumarate handle with RNF126 as well as additional RING-family E3 ligases. We then transplanted this covalent handle onto a diverse set of protein-targeting ligands to induce the degradation of BRD4, BCR-ABL and c-ABL, PDE5, AR and AR-V7, BTK, LRRK2, HDAC1/3, and SMARCA2/4. Our study undercovers a design strategy for converting protein-targeting ligands into covalent molecular glue degraders.
© 2023 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare the following competing financial interest(s): JAT, JMK, MS, DD, ACK, HS, AOF, MBS, HS, SMB, FJG, MJH, and LM are employees of Novartis Institutes for BioMedical Research. JP was a University of California, Berkeley employee during this study, but is now a Novartis Institutes for BioMedical Research employee. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Translational Chemical Biology Institute. DKN is a co-founder, shareholder, and scientific advisory board member for Frontier Medicines and Vicinitas Therapeutics. DKN is a member of the board of directors for Vicinitas Therapeutics. DKN is also on the scientific advisory board of The Mark Foundation for Cancer Research, MD Anderson Cancer Center, Photys Therapeutics, Apertor Pharmaceuticals, Oerth Bio, and Chordia Therapeutics. DKN is also an Investment Advisory Board Member for Droia Ventures and a16z.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
