

Identification of broad, potent antibodies to functionally constrained regions of SARS-CoV-2 spike following a breakthrough infection
- PMID: 37253011
- PMCID: PMC10265947
- DOI: 10.1073/pnas.2220948120
Identification of broad, potent antibodies to functionally constrained regions of SARS-CoV-2 spike following a breakthrough infection
Abstract
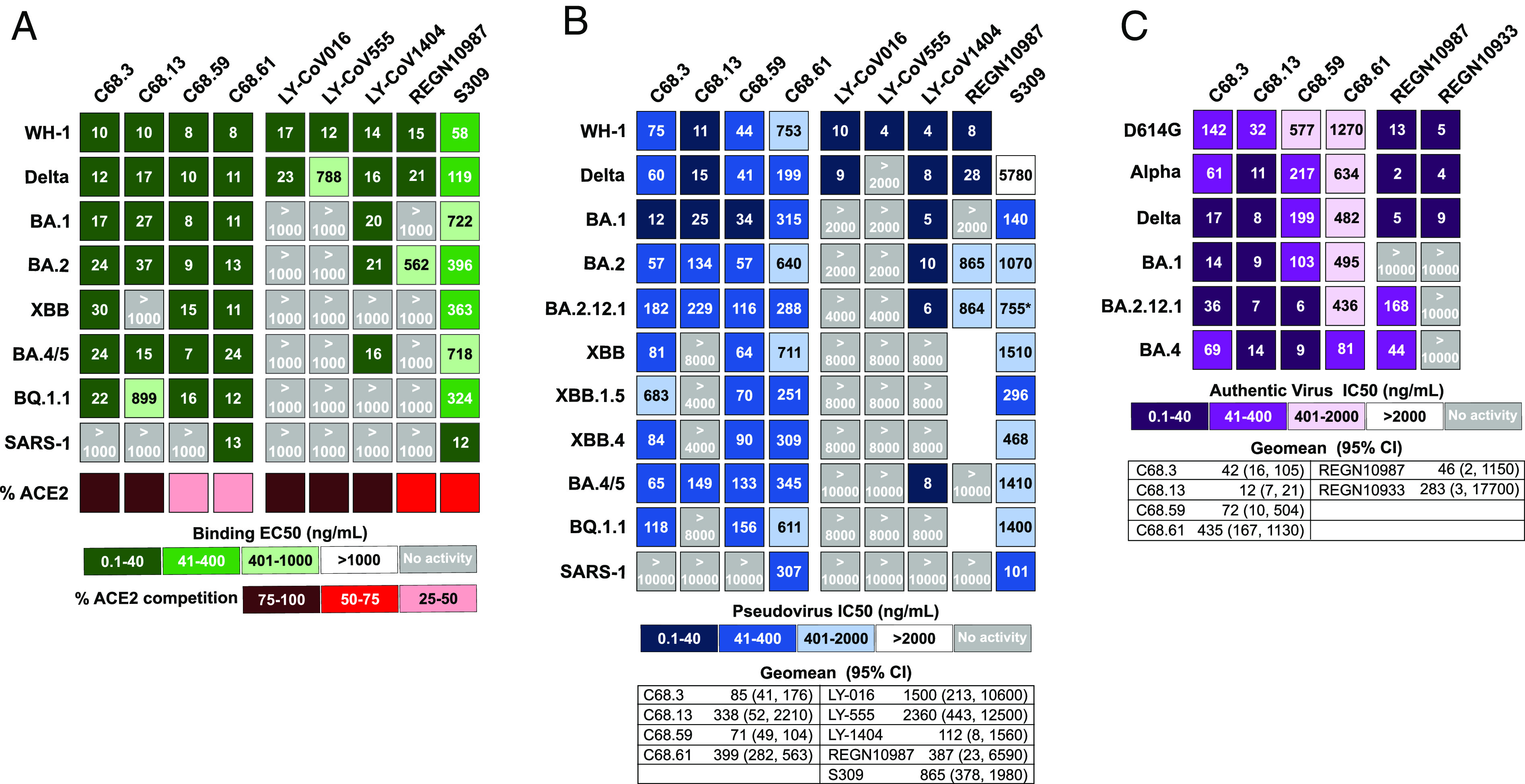
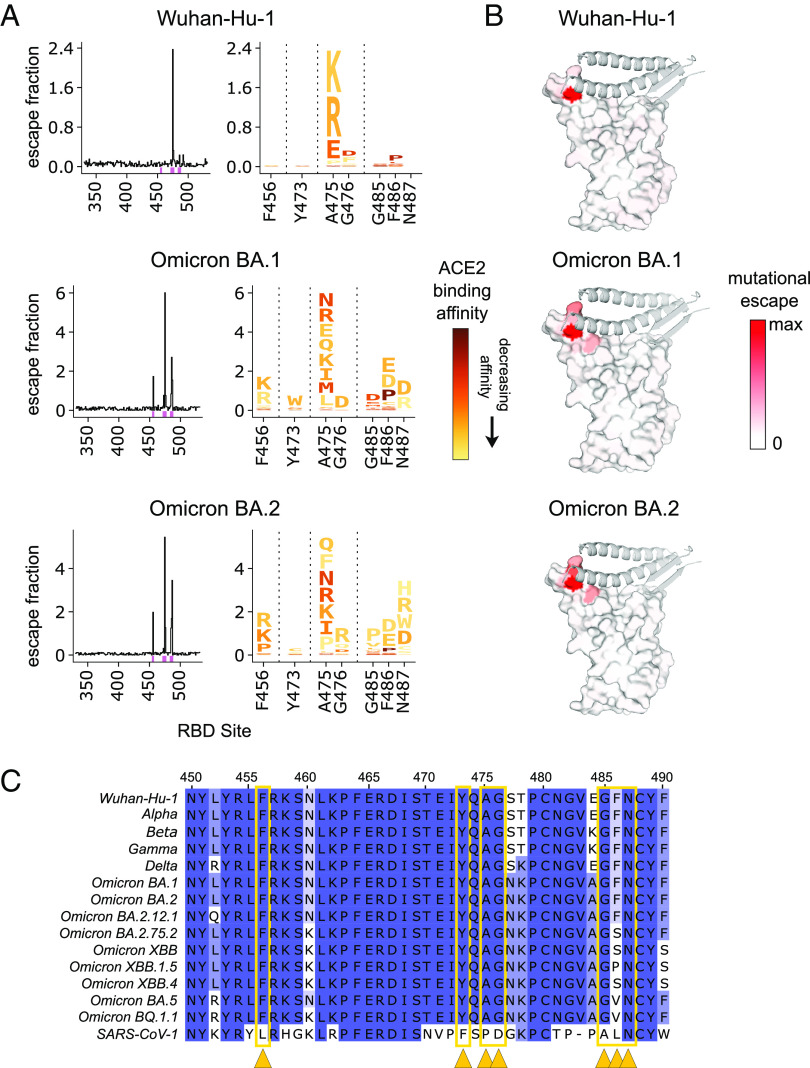
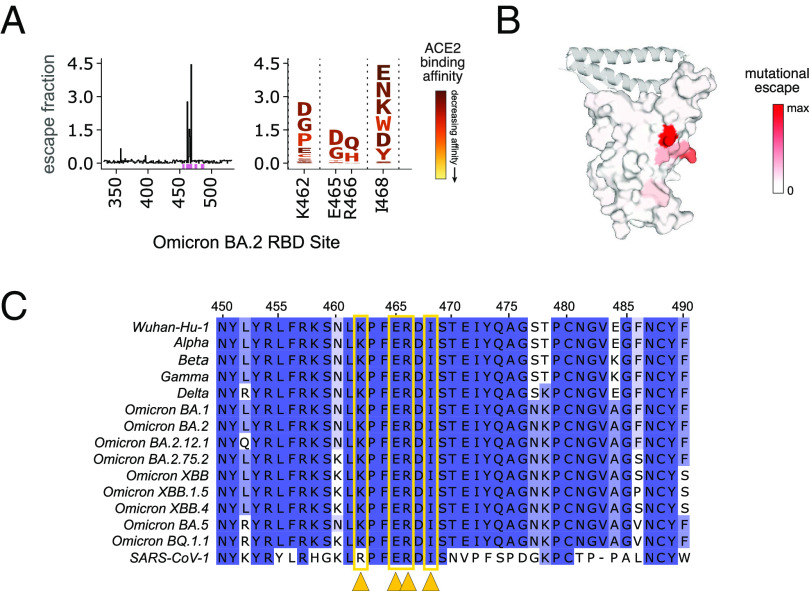
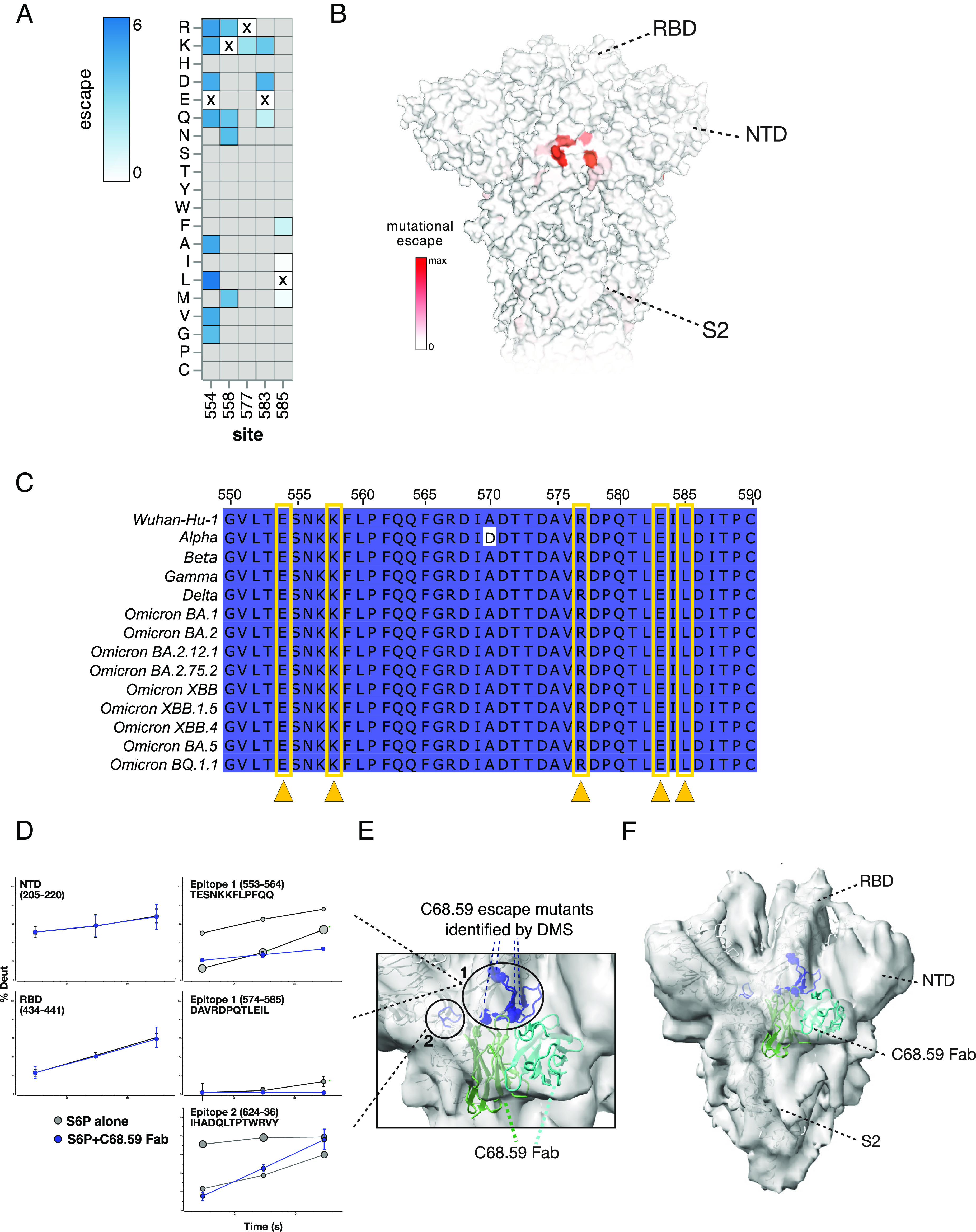
The antiviral benefit of antibodies can be compromised by viral escape especially for rapidly evolving viruses. Therefore, durable, effective antibodies must be both broad and potent to counter newly emerging, diverse strains. Discovery of such antibodies is critically important for SARS-CoV-2 as the global emergence of new variants of concern (VOC) has compromised the efficacy of therapeutic antibodies and vaccines. We describe a collection of broad and potent neutralizing monoclonal antibodies (mAbs) isolated from an individual who experienced a breakthrough infection with the Delta VOC. Four mAbs potently neutralize the Wuhan-Hu-1 vaccine strain, the Delta VOC, and also retain potency against the Omicron VOCs through BA.4/BA.5 in both pseudovirus-based and authentic virus assays. Three mAbs also retain potency to recently circulating VOCs XBB.1.5 and BQ.1.1 and one also potently neutralizes SARS-CoV-1. The potency of these mAbs was greater against Omicron VOCs than all but one of the mAbs that had been approved for therapeutic applications. The mAbs target distinct epitopes on the spike glycoprotein, three in the receptor-binding domain (RBD) and one in an invariant region downstream of the RBD in subdomain 1 (SD1). The escape pathways we defined at single amino acid resolution with deep mutational scanning show they target conserved, functionally constrained regions of the glycoprotein, suggesting escape could incur a fitness cost. Overall, these mAbs are unique in their breadth across VOCs, their epitope specificity, and include a highly potent mAb targeting a rare epitope outside of the RBD in SD1.
Keywords: SARS-CoV-2; monoclonal antibodies; spike glycoprotein; variants of concern.
Conflict of interest statement
J.O. consults for Aerium Therapeutics. T.N.S. consults for Apriori Bio on deep mutational scanning. J.D.B. serves as a scientific advisor to Apriori Bio and Oncorus. Subsequent to the completion of the research described in this manuscript, he also began to serve as a scientific advisor to Aerium Therapeutics and the Vaccine Company. H.Y.C. consults with Ellume, Merck, Abbvie, Pfizer, Medscape, Vindico, and the Bill and Melinda Gates Foundation. J.O. and J.G. are on a patent (22-173-US-PSP2) for the C68 antibodies.
Figures
Update of
-
Identification of broad, potent antibodies to functionally constrained regions of SARS-CoV-2 spike following a breakthrough infection.bioRxiv [Preprint]. 2023 Mar 29:2022.12.15.520606. doi: 10.1101/2022.12.15.520606. bioRxiv. 2023. Update in: Proc Natl Acad Sci U S A. 2023 Jun 6;120(23):e2220948120. doi: 10.1073/pnas.2220948120. PMID: 36561191 Free PMC article. Updated. Preprint.
References
-
- National Institutes of Health, SARS-CoV-2 variants and susceptibility to Anti-SARS-CoV-2 monoclonal antibodies (2022). https://www.covid19treatmentguidelines.nih.gov/tables/variants-and-susce.... Accessed 6 March 2023.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
