

Learning perturbation-inducible cell states from observability analysis of transcriptome dynamics
- PMID: 37253722
- PMCID: PMC10229592
- DOI: 10.1038/s41467-023-37897-9
Learning perturbation-inducible cell states from observability analysis of transcriptome dynamics
Erratum in
-
Author Correction: Learning perturbation-inducible cell states from observability analysis of transcriptome dynamics.Nat Commun. 2024 Mar 6;15(1):2034. doi: 10.1038/s41467-024-46433-2. Nat Commun. 2024. PMID: 38448488 Free PMC article. No abstract available.
Abstract
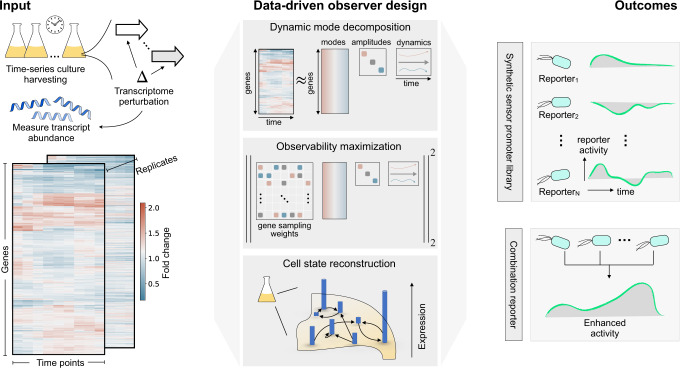
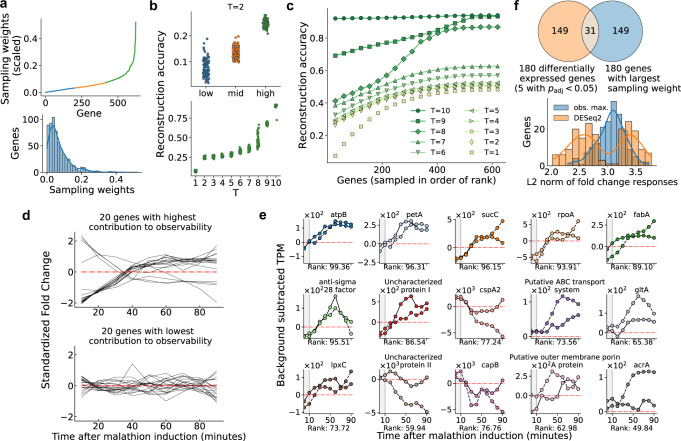
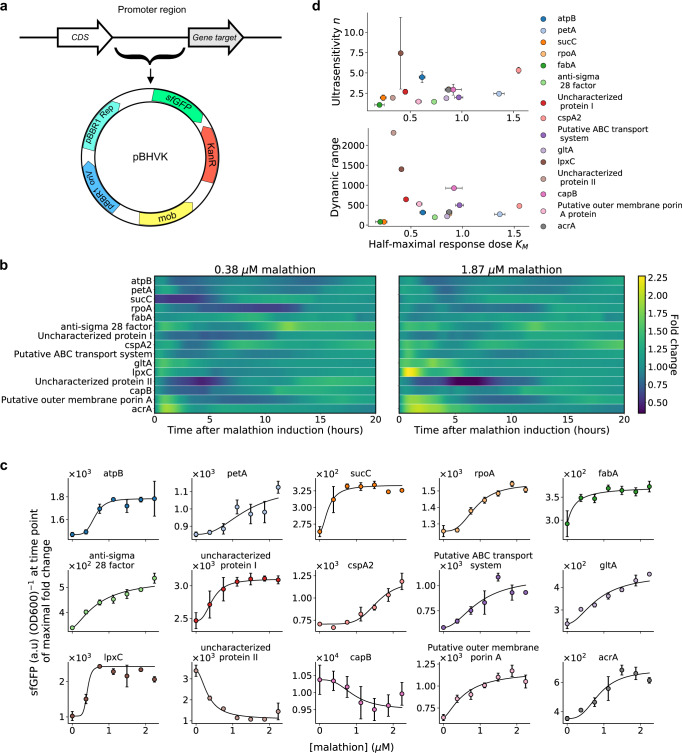
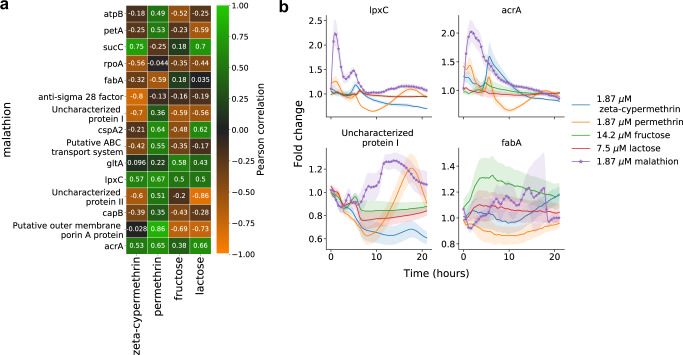
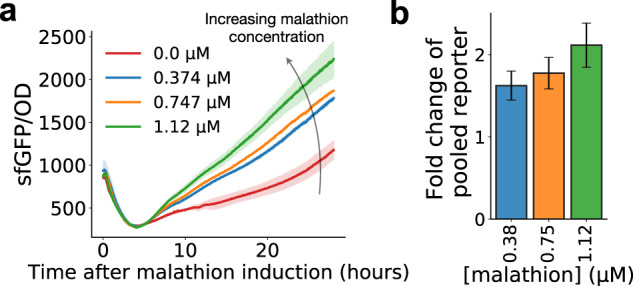
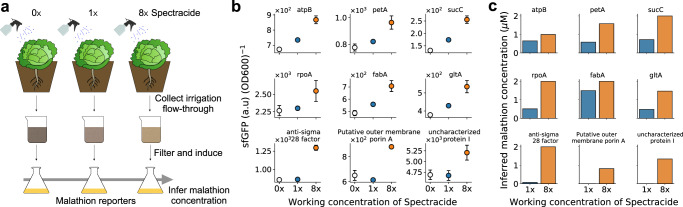
A major challenge in biotechnology and biomanufacturing is the identification of a set of biomarkers for perturbations and metabolites of interest. Here, we develop a data-driven, transcriptome-wide approach to rank perturbation-inducible genes from time-series RNA sequencing data for the discovery of analyte-responsive promoters. This provides a set of biomarkers that act as a proxy for the transcriptional state referred to as cell state. We construct low-dimensional models of gene expression dynamics and rank genes by their ability to capture the perturbation-specific cell state using a novel observability analysis. Using this ranking, we extract 15 analyte-responsive promoters for the organophosphate malathion in the underutilized host organism Pseudomonas fluorescens SBW25. We develop synthetic genetic reporters from each analyte-responsive promoter and characterize their response to malathion. Furthermore, we enhance malathion reporting through the aggregation of the response of individual reporters with a synthetic consortium approach, and we exemplify the library's ability to be useful outside the lab by detecting malathion in the environment. The engineered host cell, a living malathion sensor, can be optimized for use in environmental diagnostics while the developed machine learning tool can be applied to discover perturbation-inducible gene expression systems in the compendium of host organisms.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Differential gene expression in normal human mammary epithelial cells treated with malathion monitored by DNA microarrays.Environ Health Perspect. 2005 Aug;113(8):1046-51. doi: 10.1289/ehp.7311. Environ Health Perspect. 2005. PMID: 16079077 Free PMC article.
-
Global Regulatory Roles of the Histidine-Responsive Transcriptional Repressor HutC in Pseudomonas fluorescens SBW25.J Bacteriol. 2020 Jun 9;202(13):e00792-19. doi: 10.1128/JB.00792-19. Print 2020 Jun 9. J Bacteriol. 2020. PMID: 32291279 Free PMC article.
-
An Iterative, Synthetic Approach To Engineer a High-Performance PhoB-Specific Reporter.Appl Environ Microbiol. 2018 Jul 2;84(14):e00603-18. doi: 10.1128/AEM.00603-18. Print 2018 Jul 15. Appl Environ Microbiol. 2018. PMID: 29752265 Free PMC article.
-
Corynebacterium glutamicum promoters: a practical approach.Microb Biotechnol. 2013 Mar;6(2):103-17. doi: 10.1111/1751-7915.12019. Epub 2013 Jan 10. Microb Biotechnol. 2013. PMID: 23305350 Free PMC article. Review.
-
Advances in promoter engineering: Novel applications and predefined transcriptional control.Biotechnol J. 2021 Oct;16(10):e2100239. doi: 10.1002/biot.202100239. Epub 2021 Aug 22. Biotechnol J. 2021. PMID: 34351706 Review.
Cited by
-
Accelerating Genetic Sensor Development, Scale-up, and Deployment Using Synthetic Biology.Biodes Res. 2024 Jun 25;6:0037. doi: 10.34133/bdr.0037. eCollection 2024. Biodes Res. 2024. PMID: 38919711 Free PMC article.
-
TaRTLEt: Transcriptionally-active Riboswitch Tracer Leveraging Edge deTection.PeerJ. 2025 May 26;13:e19418. doi: 10.7717/peerj.19418. eCollection 2025. PeerJ. 2025. PMID: 40444283 Free PMC article.
-
Dynamic gene regulatory network inference from single-cell data using optimal transport.Bioinformatics. 2025 Aug 2;41(8):btaf394. doi: 10.1093/bioinformatics/btaf394. Bioinformatics. 2025. PMID: 40650986 Free PMC article.
-
Advancing reproducibility can ease the 'hard truths' of synthetic biology.Synth Biol (Oxf). 2023 Oct 28;8(1):ysad014. doi: 10.1093/synbio/ysad014. eCollection 2023. Synth Biol (Oxf). 2023. PMID: 38022744 Free PMC article. Review.
-
Predicting plant trait dynamics from genetic markers.Nat Plants. 2025 May;11(5):1018-1027. doi: 10.1038/s41477-025-01986-y. Epub 2025 Apr 17. Nat Plants. 2025. PMID: 40247143 Free PMC article.
References
-
- Bousse L. Whole cell biosensors. Sens. Actuat. B: Chem. 1996;34:270–275. doi: 10.1016/S0925-4005(96)01906-5. - DOI
-
- Song, Y. et al. Application of bacterial whole-cell biosensors in health. Handb. Cell Biosens. 945–961 (2022).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
