

ADAR1-mediated RNA editing promotes B cell lymphomagenesis
- PMID: 37255666
- PMCID: PMC10225930
- DOI: 10.1016/j.isci.2023.106864
ADAR1-mediated RNA editing promotes B cell lymphomagenesis
Abstract
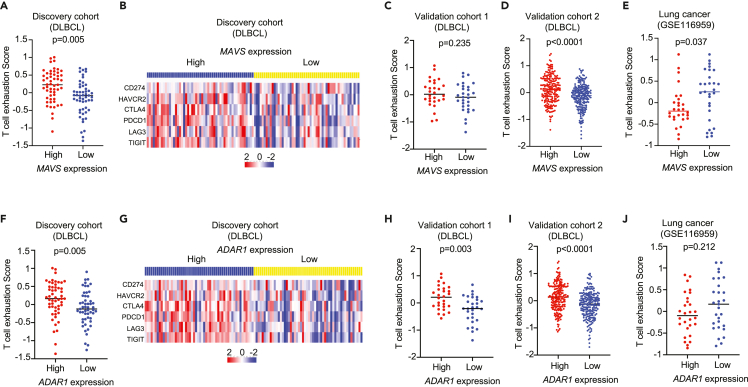
Diffuse large B cell lymphoma (DLBCL) is one of the most common types of aggressive lymphoid malignancies. Here, we explore the contribution of RNA editing to DLBCL pathogenesis. We observed that DNA mutations and RNA editing events are often mutually exclusive, suggesting that tumors can modulate pathway outcomes by altering sequences at either the genomic or the transcriptomic level. RNA editing targets transcripts within known disease-driving pathways such as apoptosis, p53 and NF-κB signaling, as well as the RIG-I-like pathway. In this context, we show that ADAR1-mediated editing within MAVS transcript positively correlates with MAVS protein expression levels, associating with increased interferon/NF-κB signaling and T cell exhaustion. Finally, using targeted RNA base editing tools to restore editing within MAVS 3'UTR in ADAR1-deficient cells, we demonstrate that editing is likely to be causal to an increase in downstream signaling in the absence of activation by canonical nucleic acid receptor sensing.
Keywords: Epigenetics; Genetics; Molecular biology; Omics; Transcriptomics.
© 2023 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
