

Cell surface marker-based capture of neoantigen-reactive CD8+ T-cell receptors from metastatic tumor digests
- PMID: 37258038
- PMCID: PMC10255266
- DOI: 10.1136/jitc-2022-006264
Cell surface marker-based capture of neoantigen-reactive CD8+ T-cell receptors from metastatic tumor digests
Abstract
Background: Cellular immunotherapies using autologous tumor-infiltrating lymphocytes (TIL) can induce durable regression of epithelial cancers in selected patients with treatment-refractory metastatic disease. As the genetic engineering of T cells with tumor-reactive T-cell receptors (TCRs) comes to the forefront of clinical investigation, the rapid, scalable, and cost-effective detection of patient-specific neoantigen-reactive TIL remains a top priority.
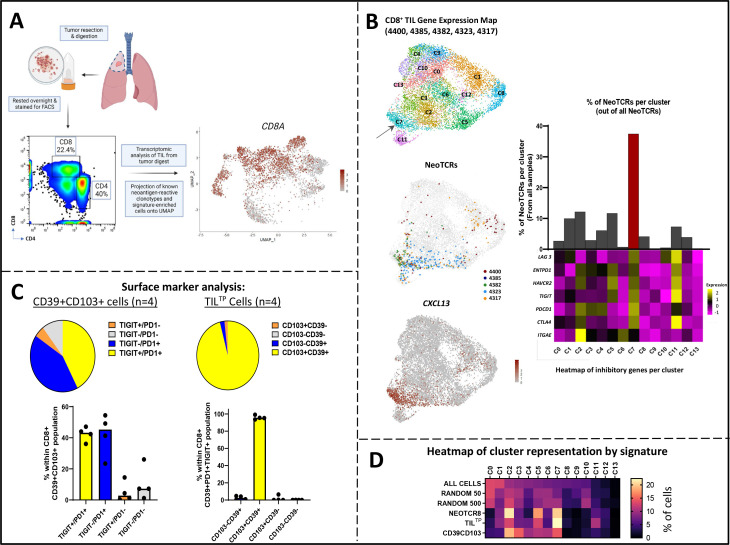
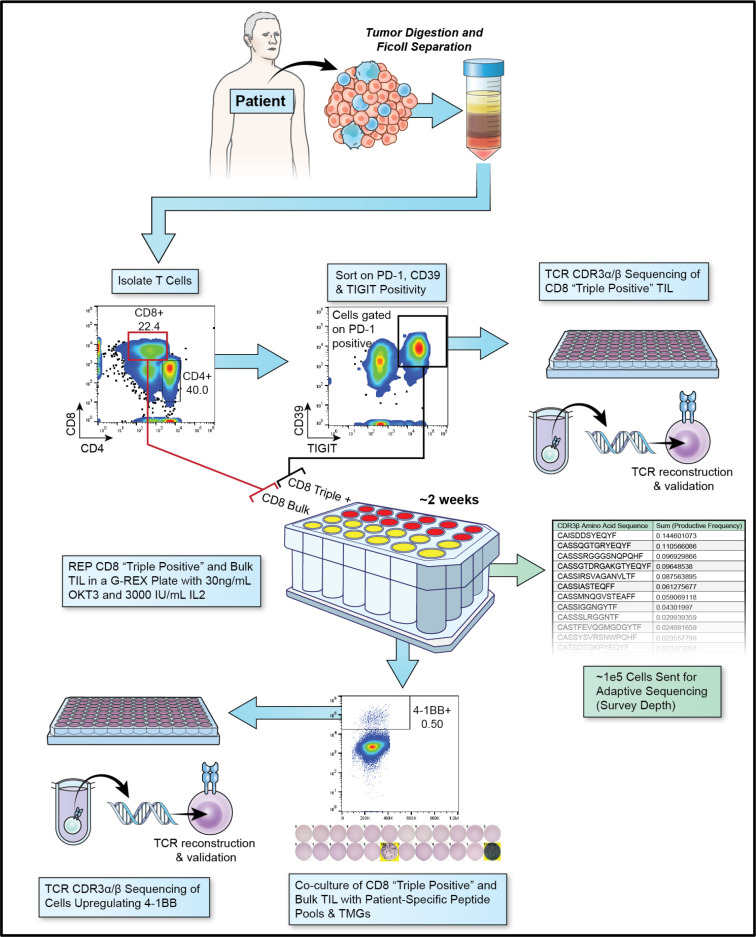
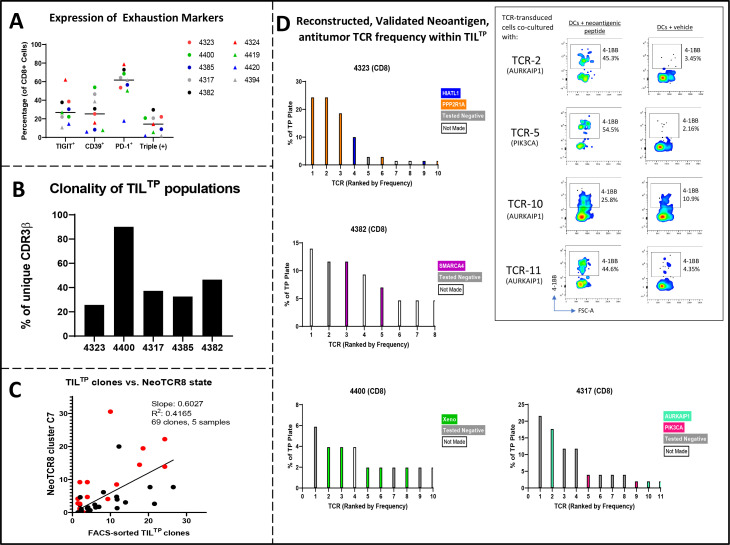
Methods: We analyzed the single-cell transcriptomic states of 31 neoantigen-specific T-cell clonotypes to identify cell surface dysfunction markers that best identified the metastatic transcriptional states enriched with antitumor TIL. We developed an efficient method to capture neoantigen-reactive TCRs directly from resected human tumors based on cell surface co-expression of CD39, programmed cell death protein-1, and TIGIT dysfunction markers (CD8+ TILTP).
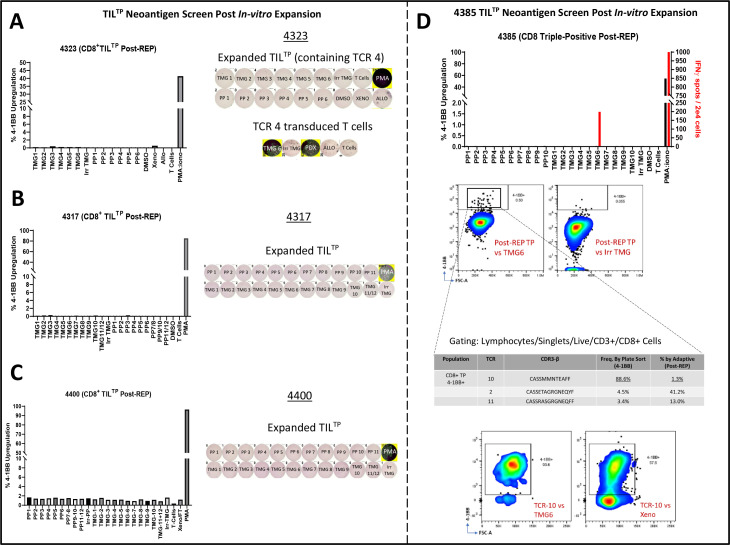
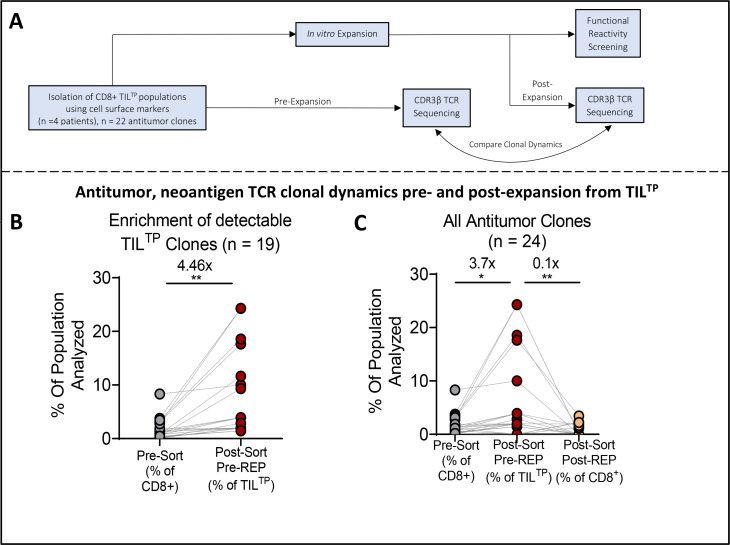
Results: TILTP TCR isolation achieved a high degree of correlation with single-cell transcriptomic signatures that identify neoantigen-reactive TCRs, making it a cost-effective strategy using widely available resources. Reconstruction of additional TILTP TCRs from tumors identified known and novel antitumor TCRs, showing that at least 39.5% of TILTP TCRs are neoantigen-reactive or tumor-reactive. Despite their substantial enrichment for neoantigen-reactive TCR clonotypes, clonal dynamics of 24 unique antitumor TILTP clonotypes from four patients indicated that most in vitro expanded TILTP populations failed to demonstrate neoantigen reactivity, either by loss of neoantigen-reactive clones during TIL expansion, or through functional impairment during cognate neoantigen recognition.
Conclusions: While direct usage of in vitro-expanded CD8+ TILTP as a source for cellular therapy might be precluded by profound TIL dysfunction, isolating TILTP represents a streamlined effective approach to rapidly identify neoantigen-reactive TCRs to design engineered cellular immunotherapies against cancer.
Keywords: CD8-Positive T-Lymphocytes; Cell Engineering; Immunity, Cellular; Immunotherapy; Lymphocytes, Tumor-Infiltrating.
© Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: FJL, SK, PR, and SAR are listed on an international patent application filed based on the NeoTCR8 signature described in this study. All other authors declare no competing interests.
Figures
References
-
- Parkhurst MR, Robbins PF, Tran E, et al. . Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov 2019;9:1022–35. 10.1158/2159-8290.CD-18-1494 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials