

Pan-KRAS inhibitor disables oncogenic signalling and tumour growth
- PMID: 37258666
- PMCID: PMC10322706
- DOI: 10.1038/s41586-023-06123-3
Pan-KRAS inhibitor disables oncogenic signalling and tumour growth
Abstract
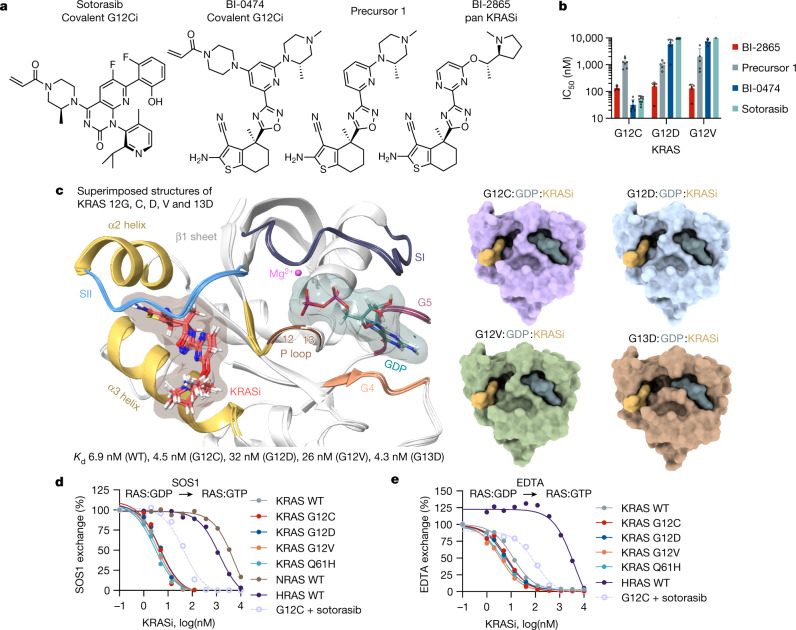
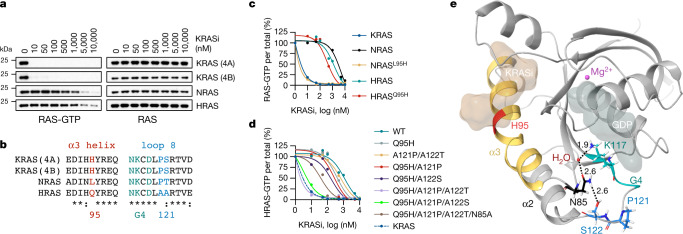
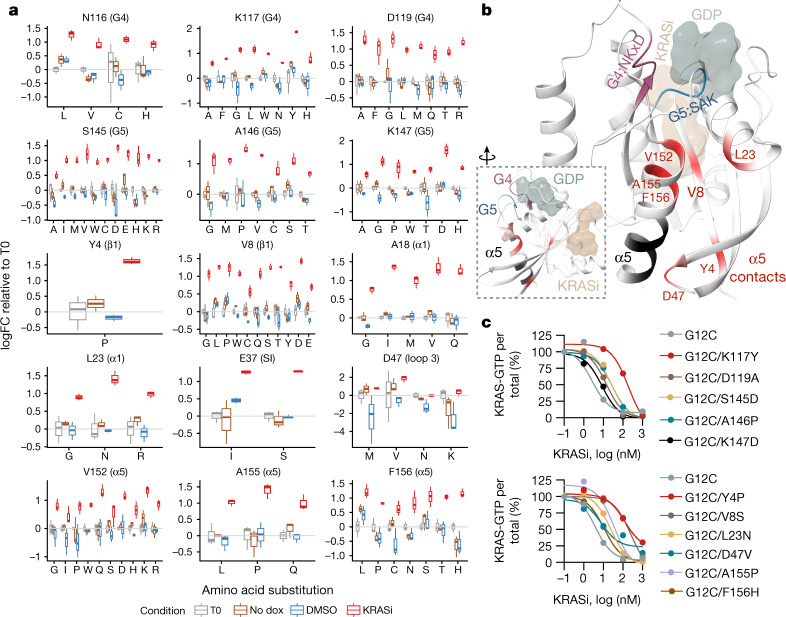
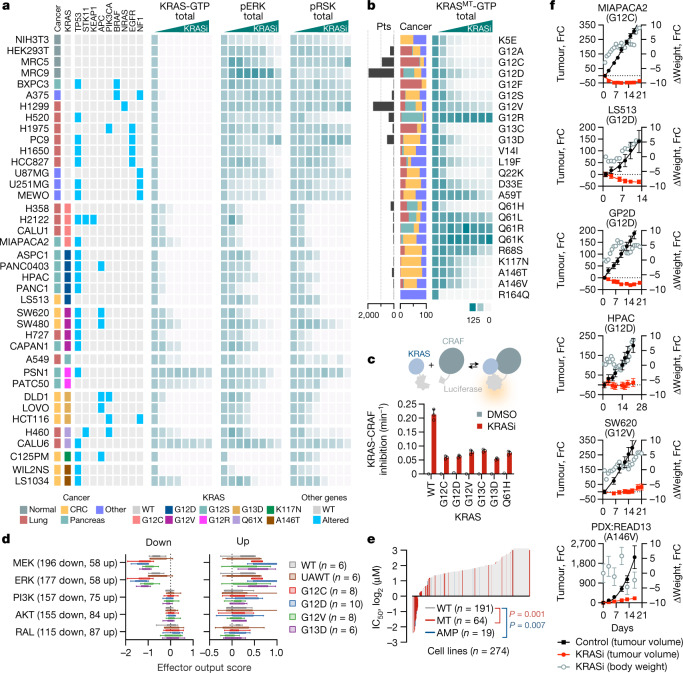
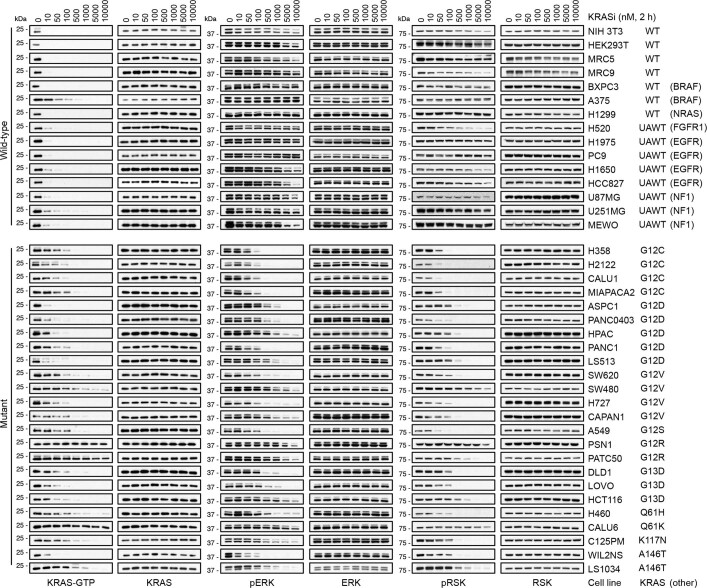
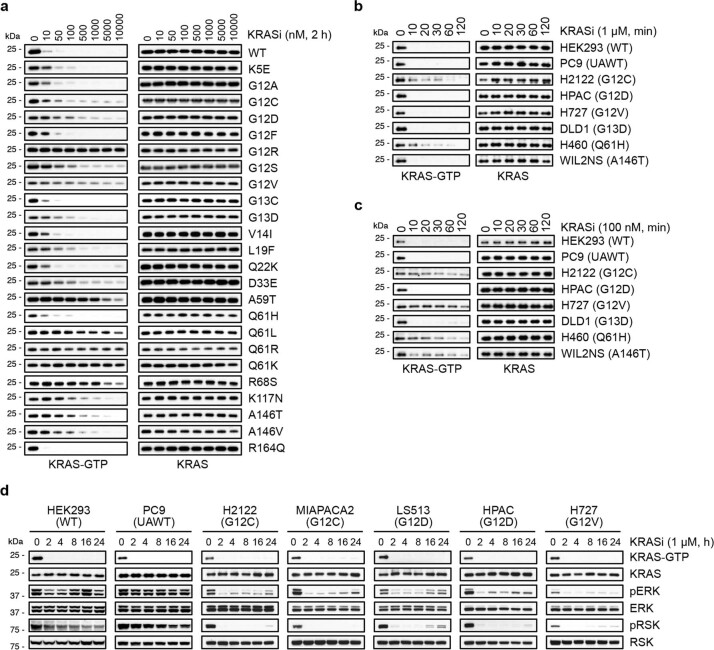
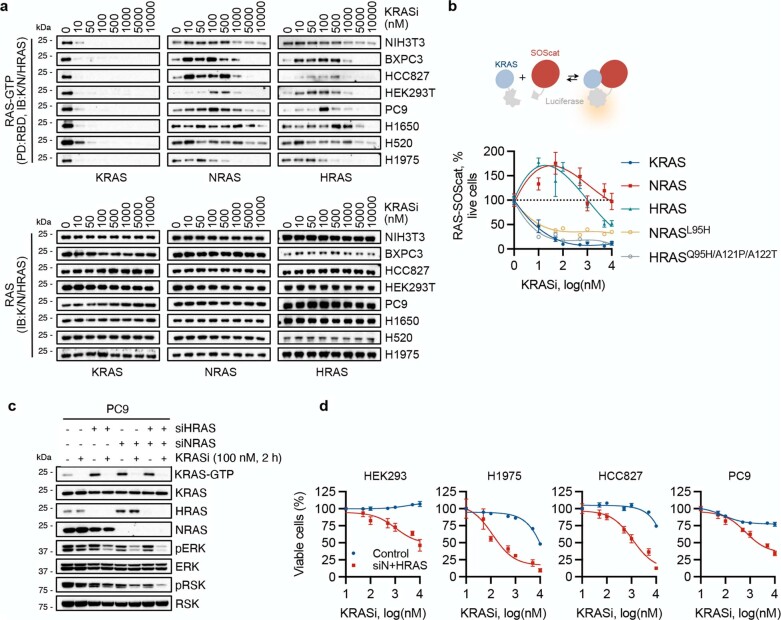
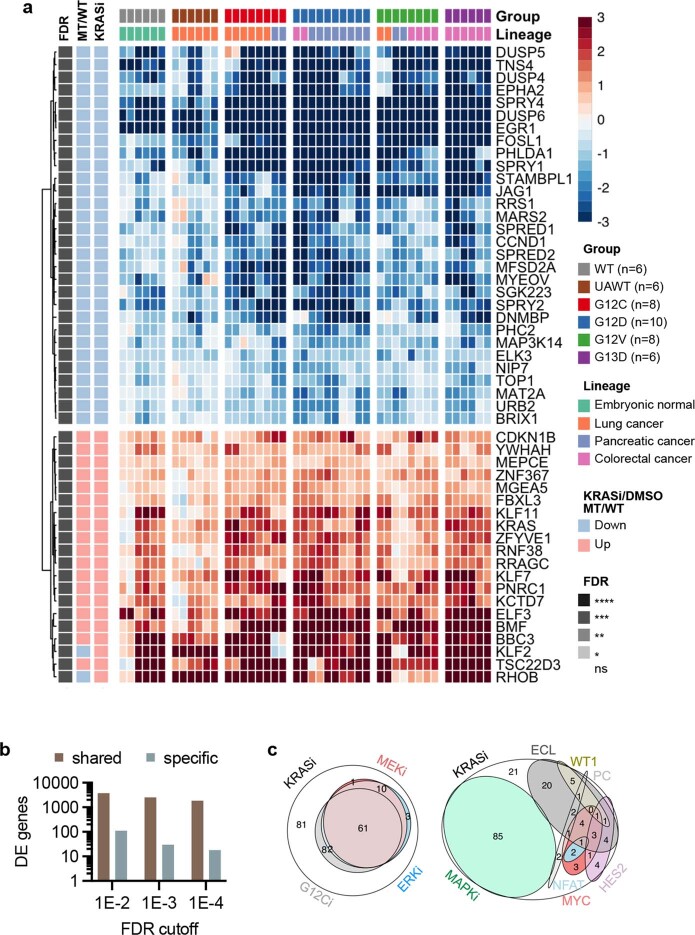
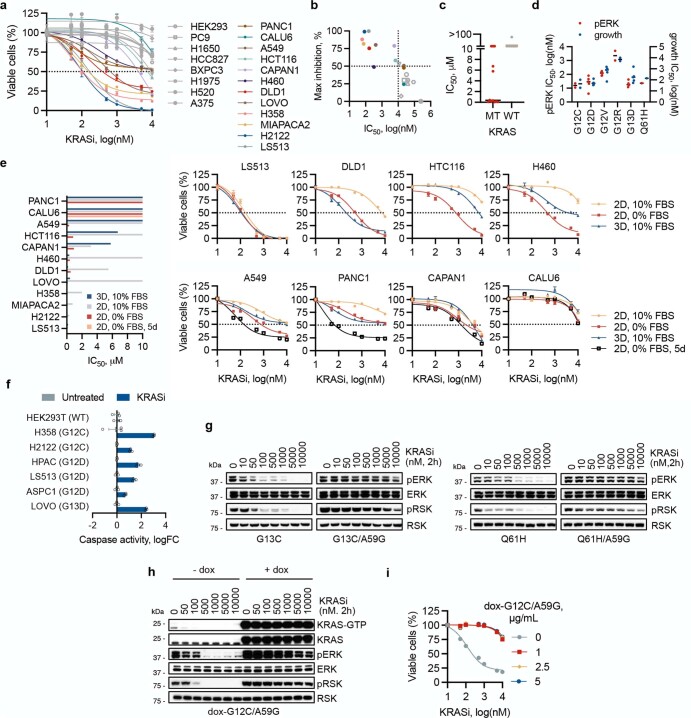
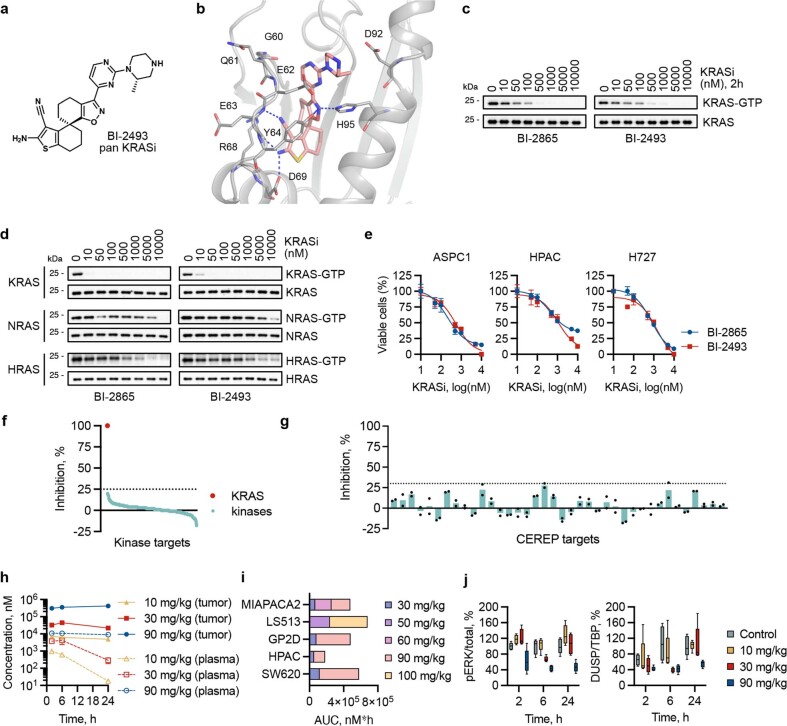
KRAS is one of the most commonly mutated proteins in cancer, and efforts to directly inhibit its function have been continuing for decades. The most successful of these has been the development of covalent allele-specific inhibitors that trap KRAS G12C in its inactive conformation and suppress tumour growth in patients1-7. Whether inactive-state selective inhibition can be used to therapeutically target non-G12C KRAS mutants remains under investigation. Here we report the discovery and characterization of a non-covalent inhibitor that binds preferentially and with high affinity to the inactive state of KRAS while sparing NRAS and HRAS. Although limited to only a few amino acids, the evolutionary divergence in the GTPase domain of RAS isoforms was sufficient to impart orthosteric and allosteric constraints for KRAS selectivity. The inhibitor blocked nucleotide exchange to prevent the activation of wild-type KRAS and a broad range of KRAS mutants, including G12A/C/D/F/V/S, G13C/D, V14I, L19F, Q22K, D33E, Q61H, K117N and A146V/T. Inhibition of downstream signalling and proliferation was restricted to cancer cells harbouring mutant KRAS, and drug treatment suppressed KRAS mutant tumour growth in mice, without having a detrimental effect on animal weight. Our study suggests that most KRAS oncoproteins cycle between an active state and an inactive state in cancer cells and are dependent on nucleotide exchange for activation. Pan-KRAS inhibitors, such as the one described here, have broad therapeutic implications and merit clinical investigation in patients with KRAS-driven cancers.
© 2023. The Author(s).
Conflict of interest statement
P.L. is listed as an inventor on patents filed by MSKCC on the treatment of KRAS or BRAF mutant cancers (US20200009138A1). P.L. reports grants to his institution from Amgen, Mirati, Revolution Medicines, Boehringer Ingelheim and Virtec Pharmaceuticals. P.L. reports consulting fees from Black Diamond Therapeutics, AmMax, OrbiMed, PAQTx, Repare Therapeutics and Revolution Medicines, travel reimbursement from Boehringer Ingelheim as well as membership of the Scientific Advisory Board of Frontier Medicines (consulting fees and equity). L.H., D.R., J.B., B.W., K.G.K., D.B.M. and N.K. are employed by Boehringer Ingelheim. The remaining authors declare no competing interests.
Figures
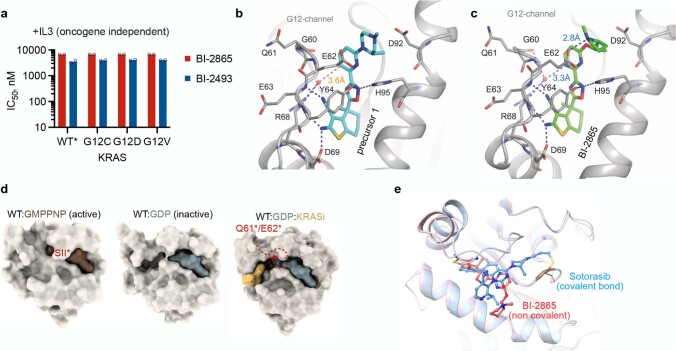
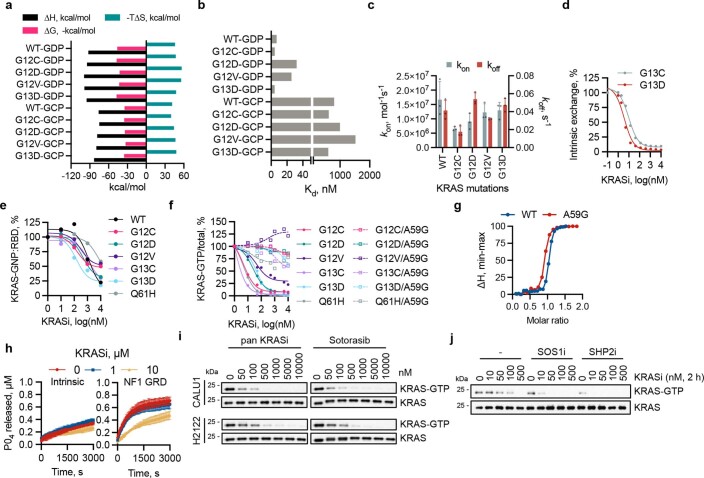
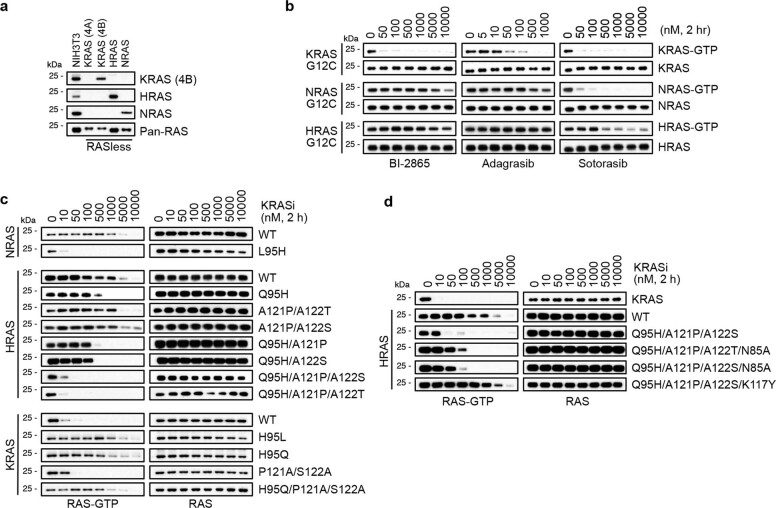
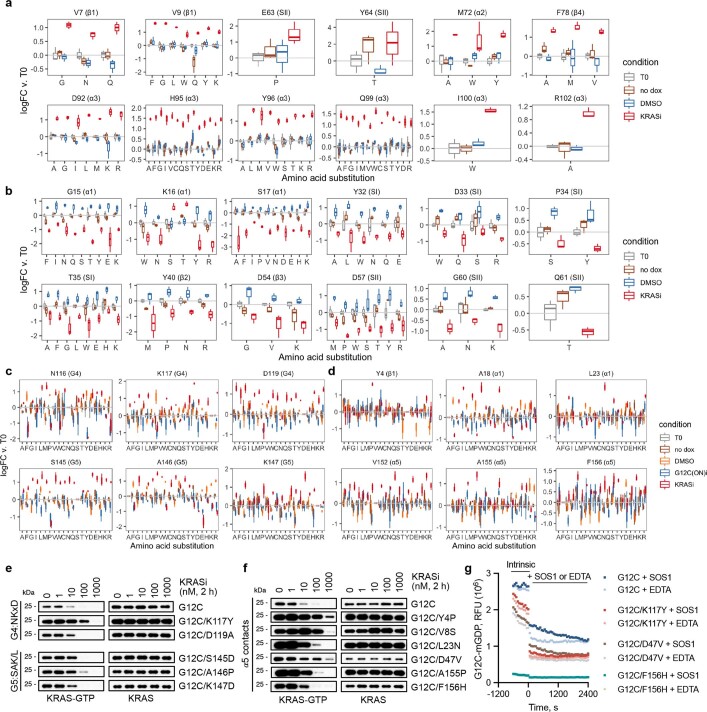
Comment in
-
A Pan-KRAS Inhibitor Impedes Common KRAS Oncoproteins and Tumor Growth.Cancer Discov. 2023 Aug 4;13(8):1757. doi: 10.1158/2159-8290.CD-RW2023-090. Cancer Discov. 2023. PMID: 37294122
-
A non-covalent inhibitor with pan-KRAS potential.Nat Rev Drug Discov. 2023 Aug;22(8):622. doi: 10.1038/d41573-023-00115-8. Nat Rev Drug Discov. 2023. PMID: 37414888 No abstract available.
References
-
- Hallin J, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
