

Transfer learning enables predictions in network biology
- PMID: 37258680
- PMCID: PMC10949956
- DOI: 10.1038/s41586-023-06139-9
Transfer learning enables predictions in network biology
Abstract
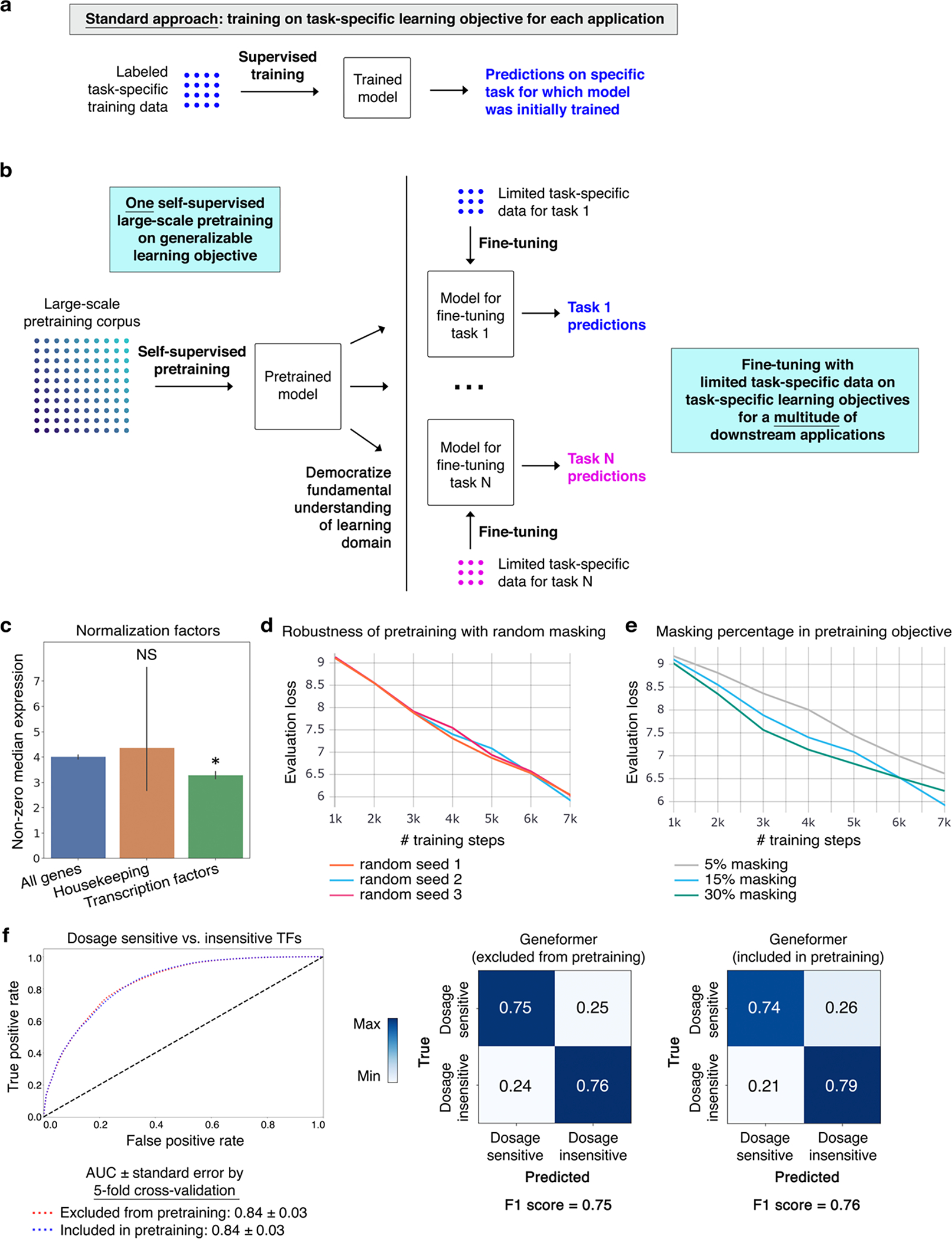
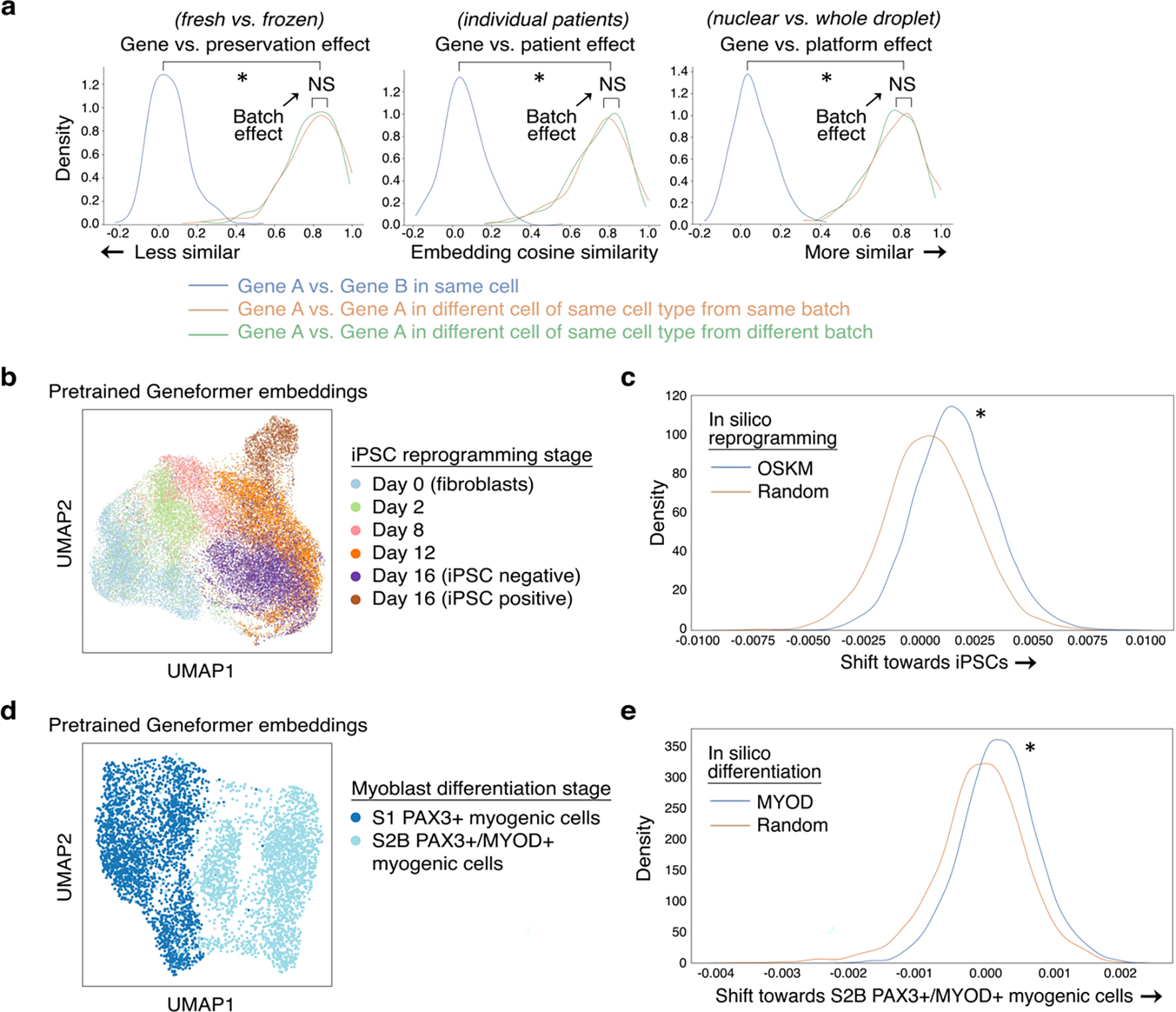
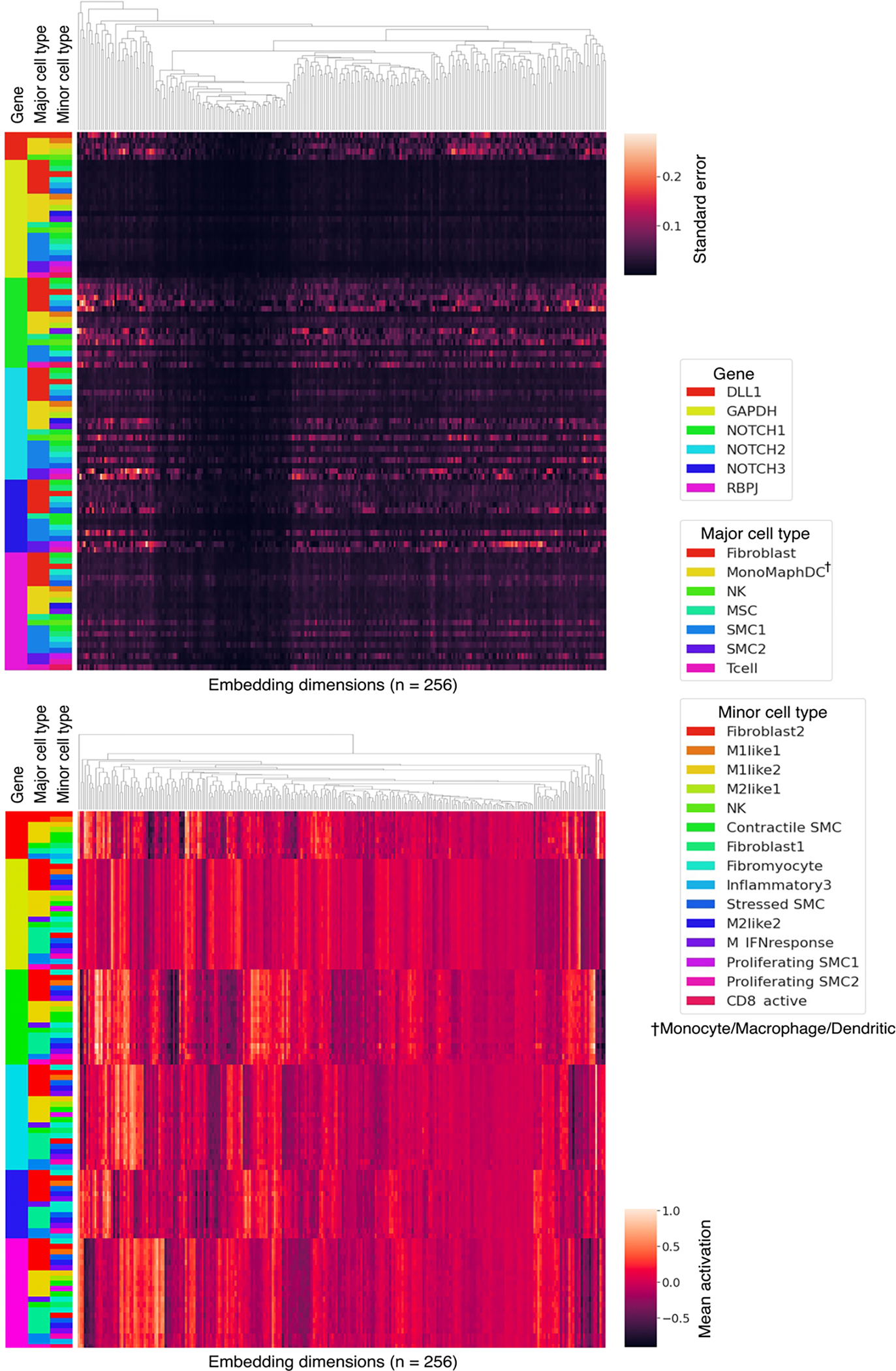
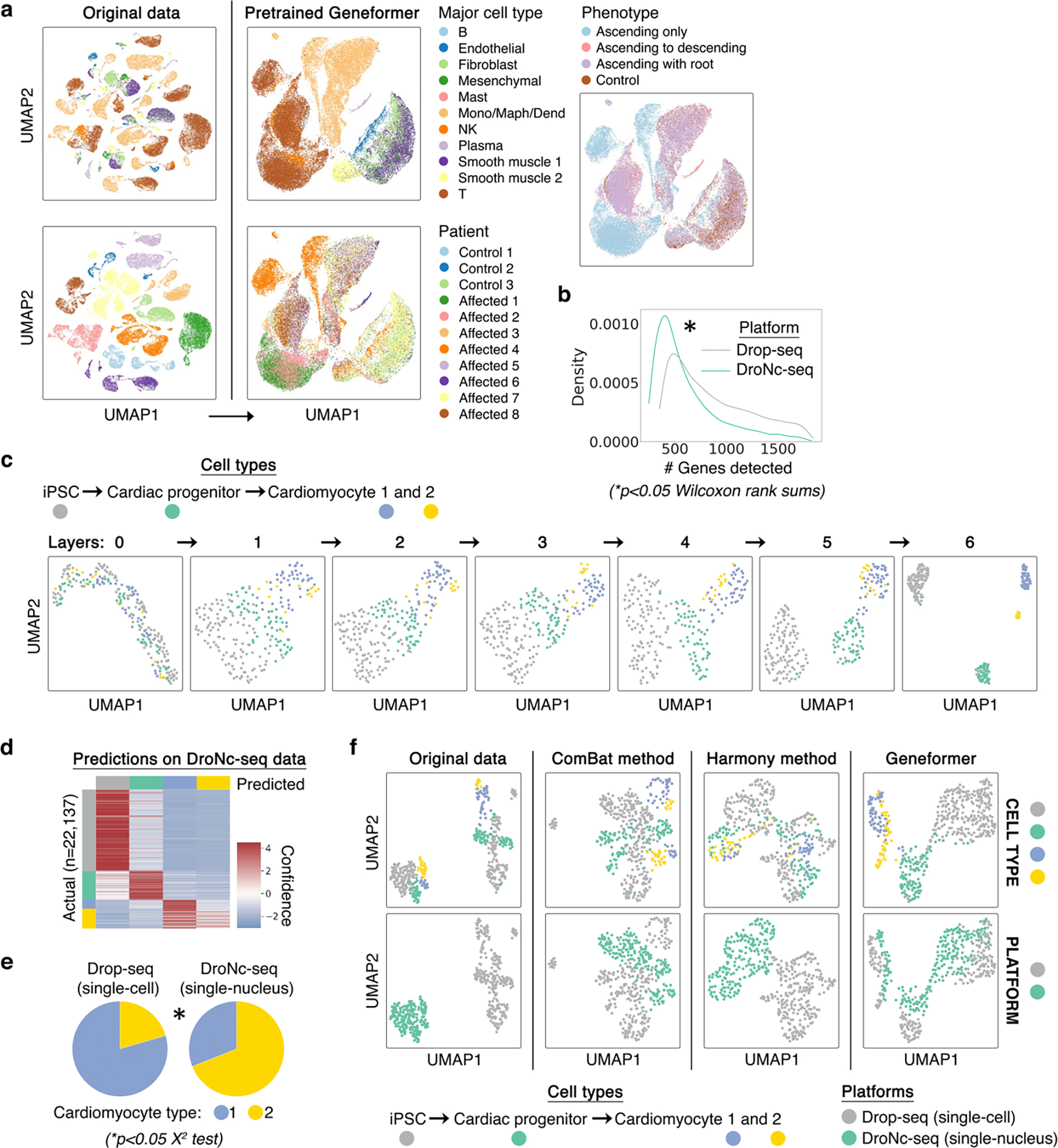
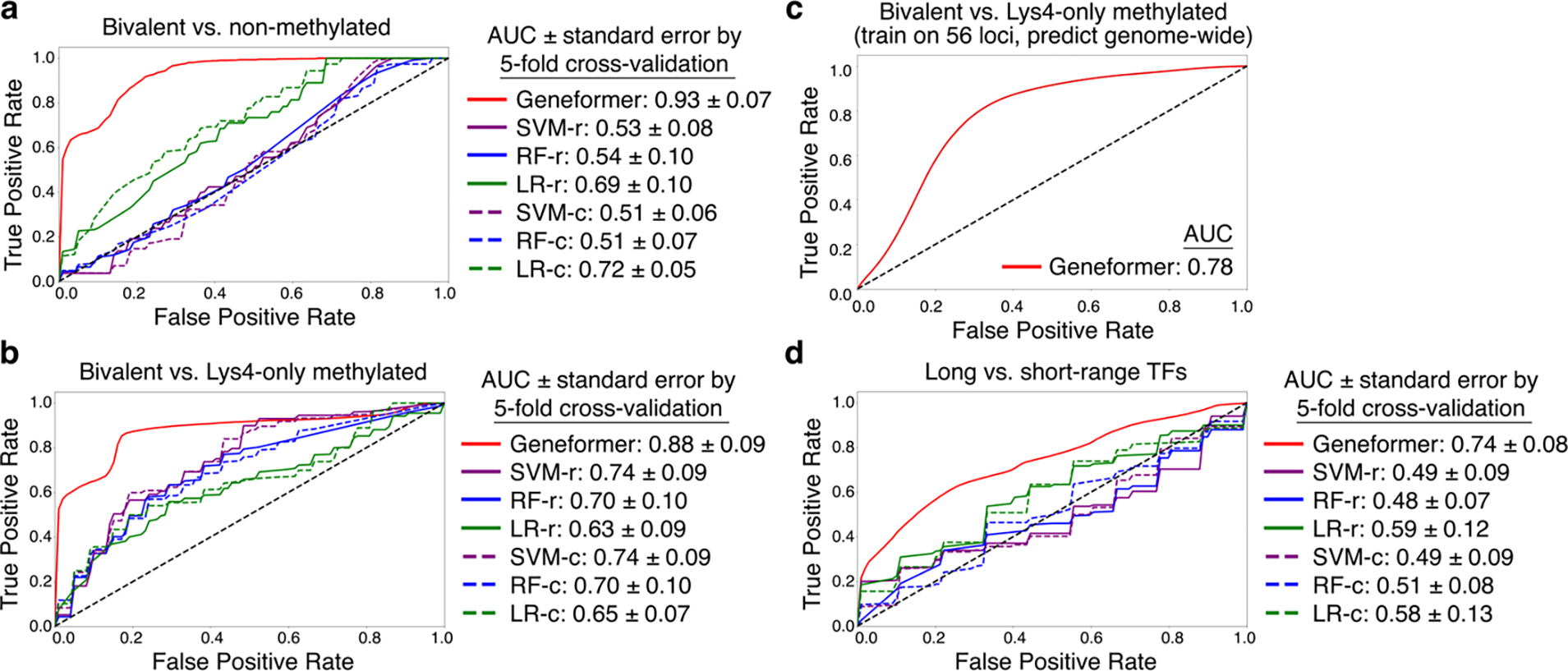
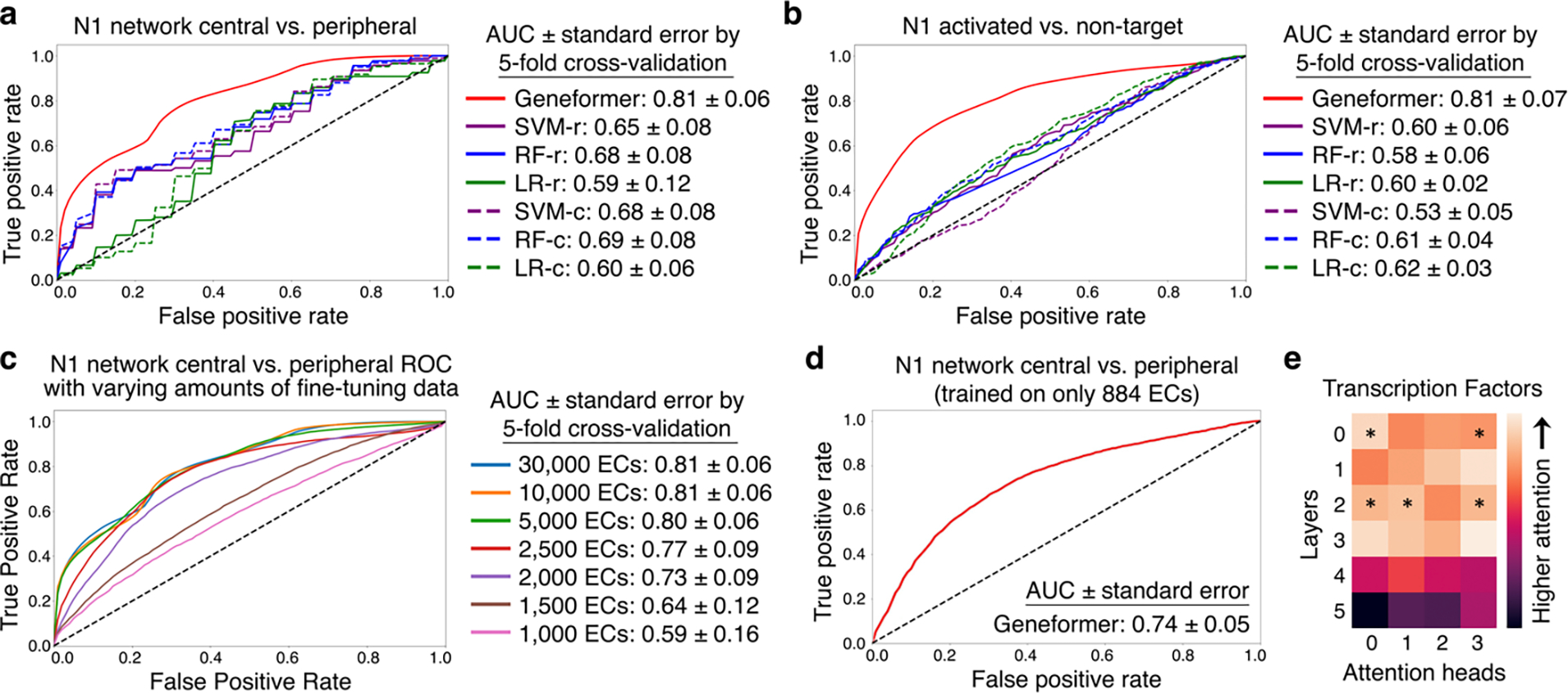
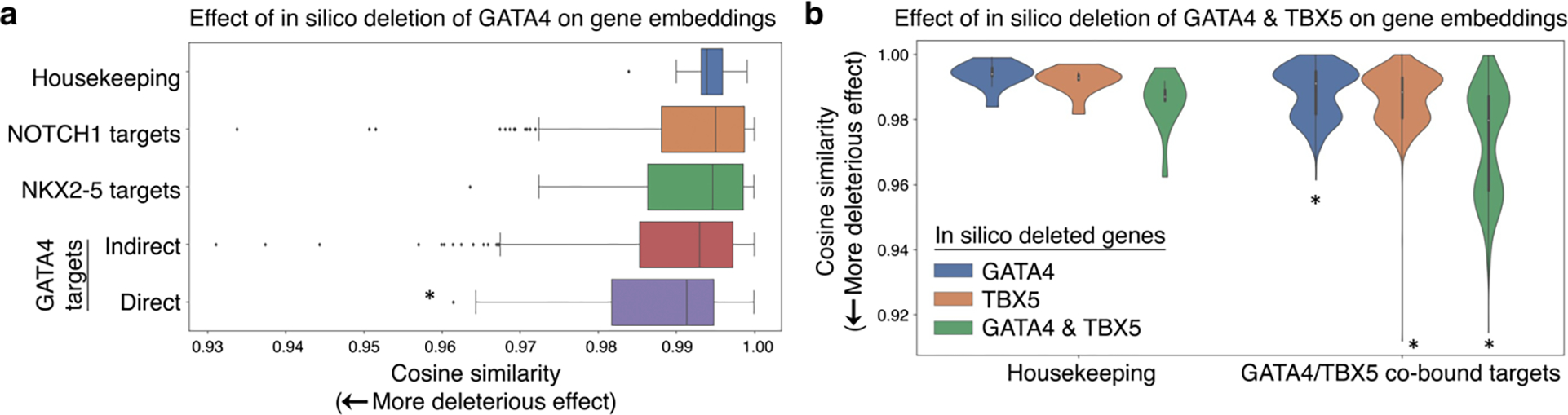
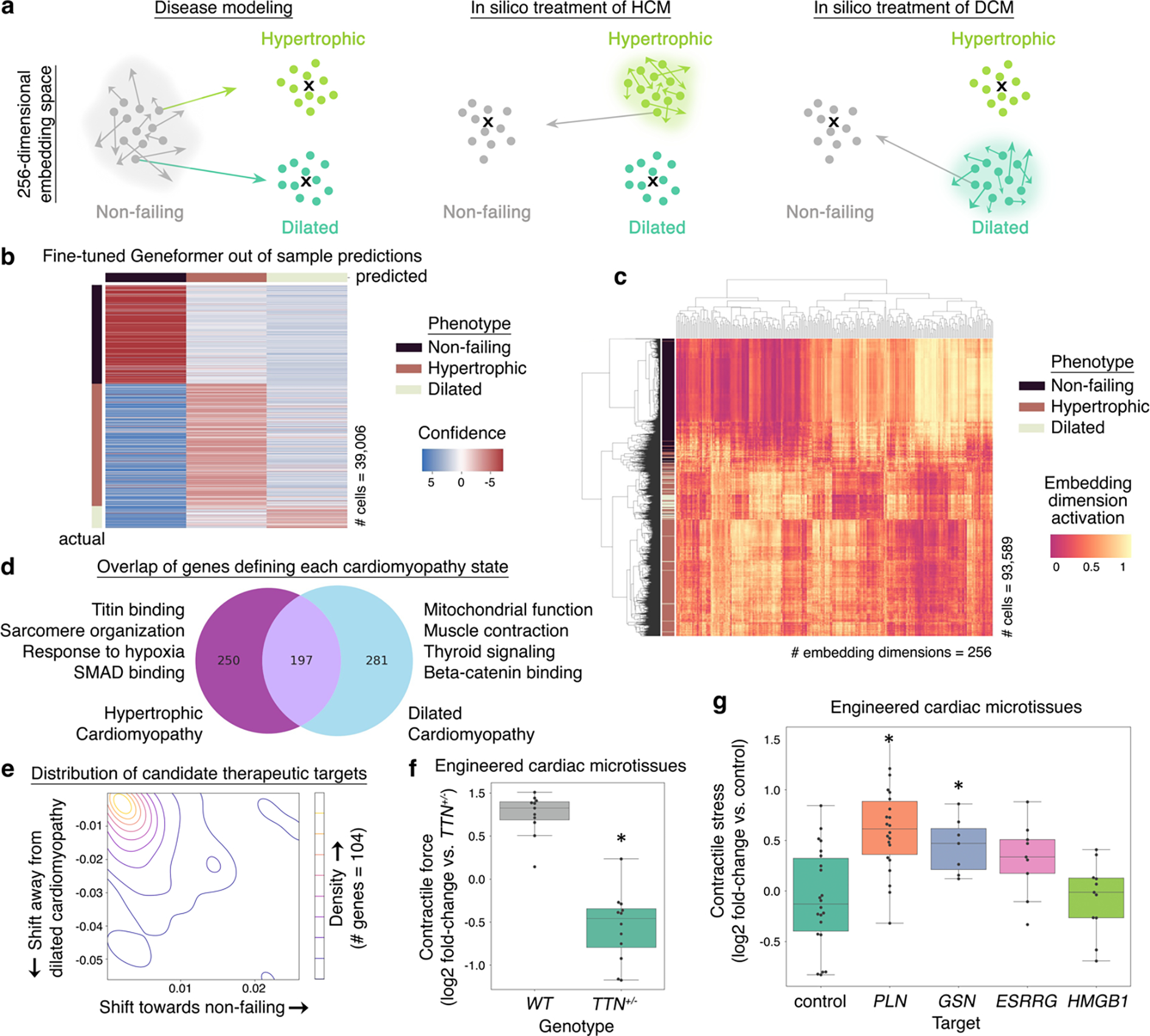
Mapping gene networks requires large amounts of transcriptomic data to learn the connections between genes, which impedes discoveries in settings with limited data, including rare diseases and diseases affecting clinically inaccessible tissues. Recently, transfer learning has revolutionized fields such as natural language understanding1,2 and computer vision3 by leveraging deep learning models pretrained on large-scale general datasets that can then be fine-tuned towards a vast array of downstream tasks with limited task-specific data. Here, we developed a context-aware, attention-based deep learning model, Geneformer, pretrained on a large-scale corpus of about 30 million single-cell transcriptomes to enable context-specific predictions in settings with limited data in network biology. During pretraining, Geneformer gained a fundamental understanding of network dynamics, encoding network hierarchy in the attention weights of the model in a completely self-supervised manner. Fine-tuning towards a diverse panel of downstream tasks relevant to chromatin and network dynamics using limited task-specific data demonstrated that Geneformer consistently boosted predictive accuracy. Applied to disease modelling with limited patient data, Geneformer identified candidate therapeutic targets for cardiomyopathy. Overall, Geneformer represents a pretrained deep learning model from which fine-tuning towards a broad range of downstream applications can be pursued to accelerate discovery of key network regulators and candidate therapeutic targets.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing Interests
XSL conducted this work while on faculty at Dana-Farber Cancer Institute and is currently a board member and CEO of GV20 Oncotherapy. PTE has received sponsored research support from Bayer AG, IBM Research, Bristol Myers Squibb, and Pfizer. PTE has also served on advisory boards or consulted for Bayer AG, MyoKardia, and Novartis. AC is an employee of Bayer US LLC (a subsidiary of Bayer AG) and may own stock in Bayer AG. EMB was a full-time employee of Bayer when this work was performed.
Figures
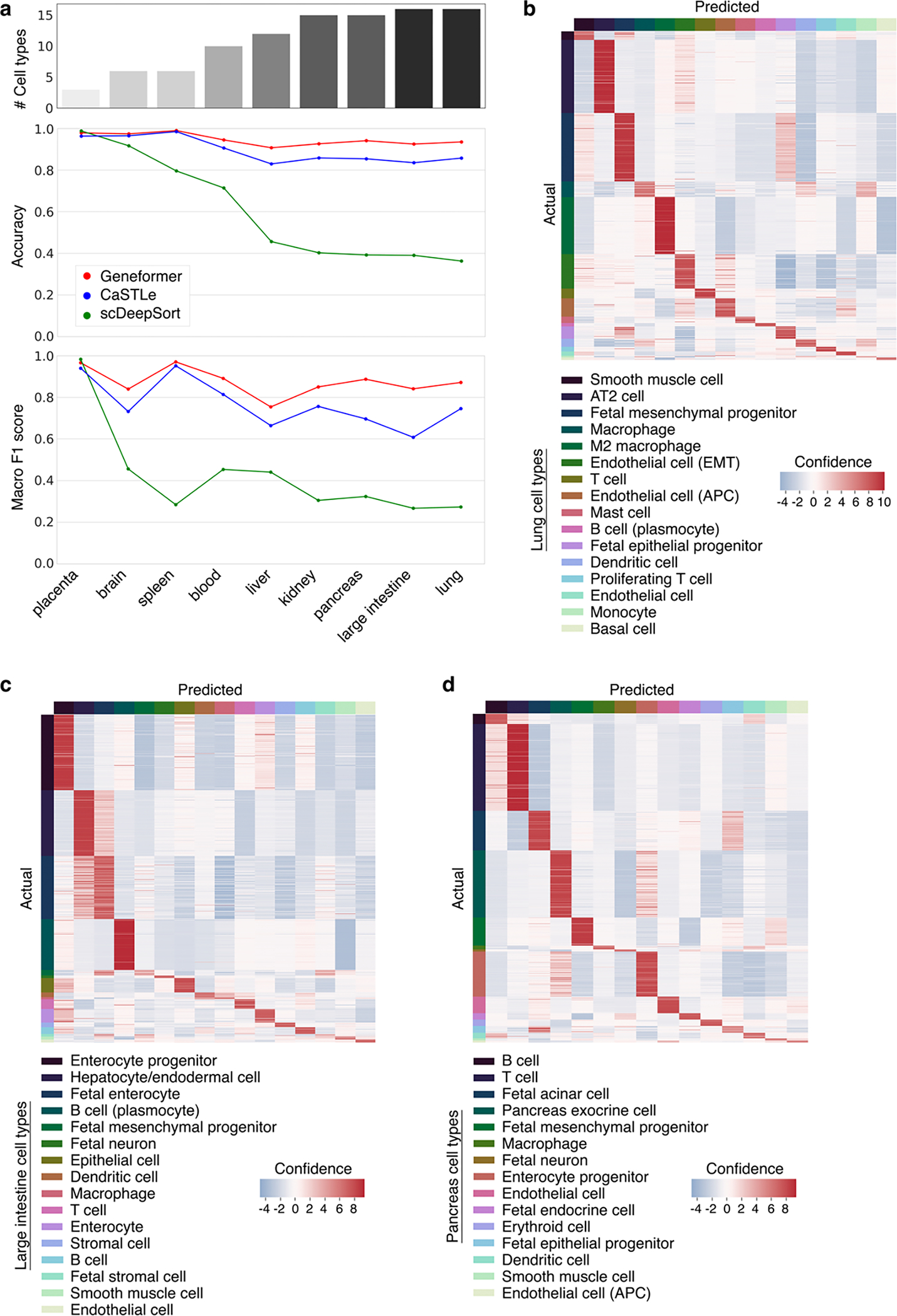
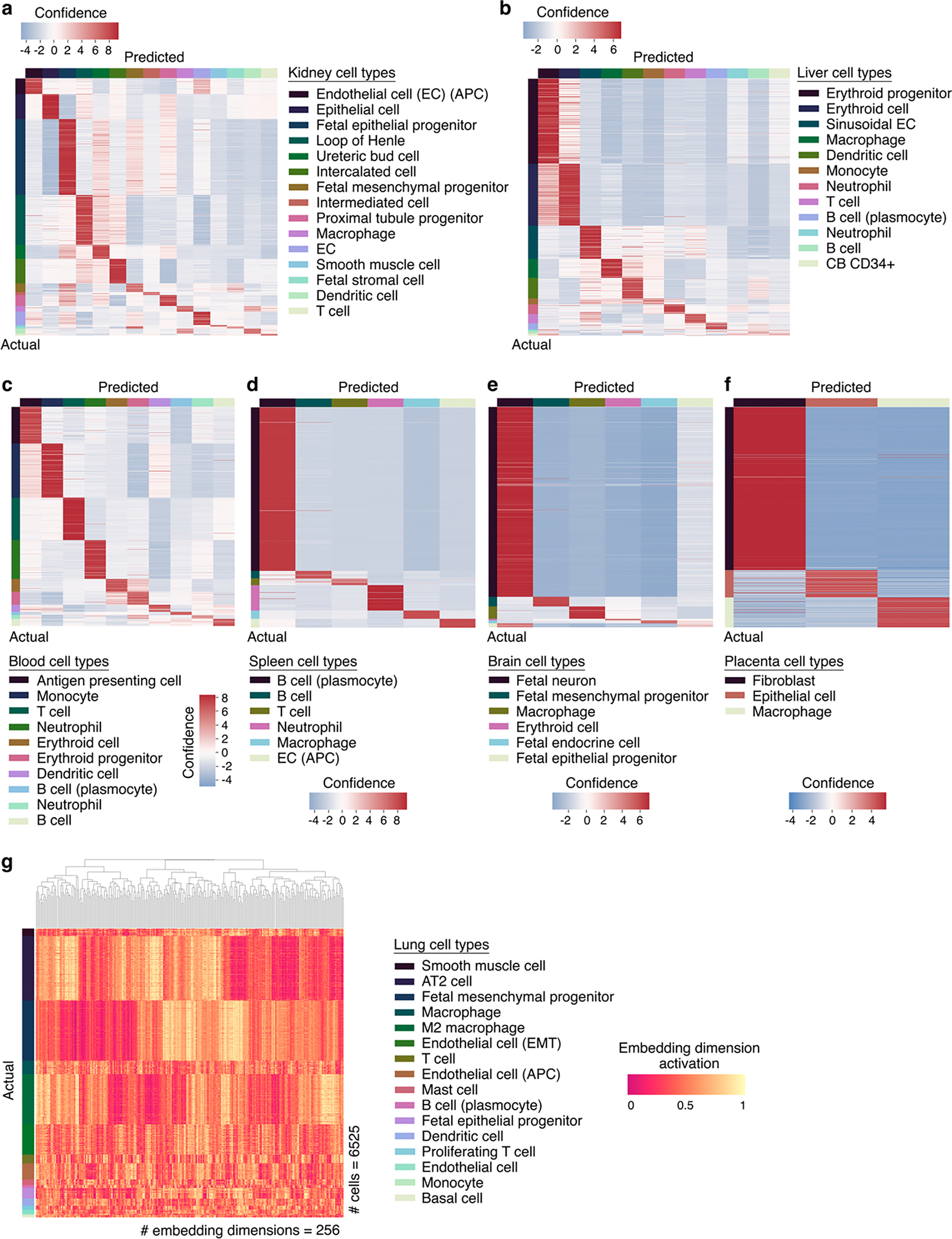
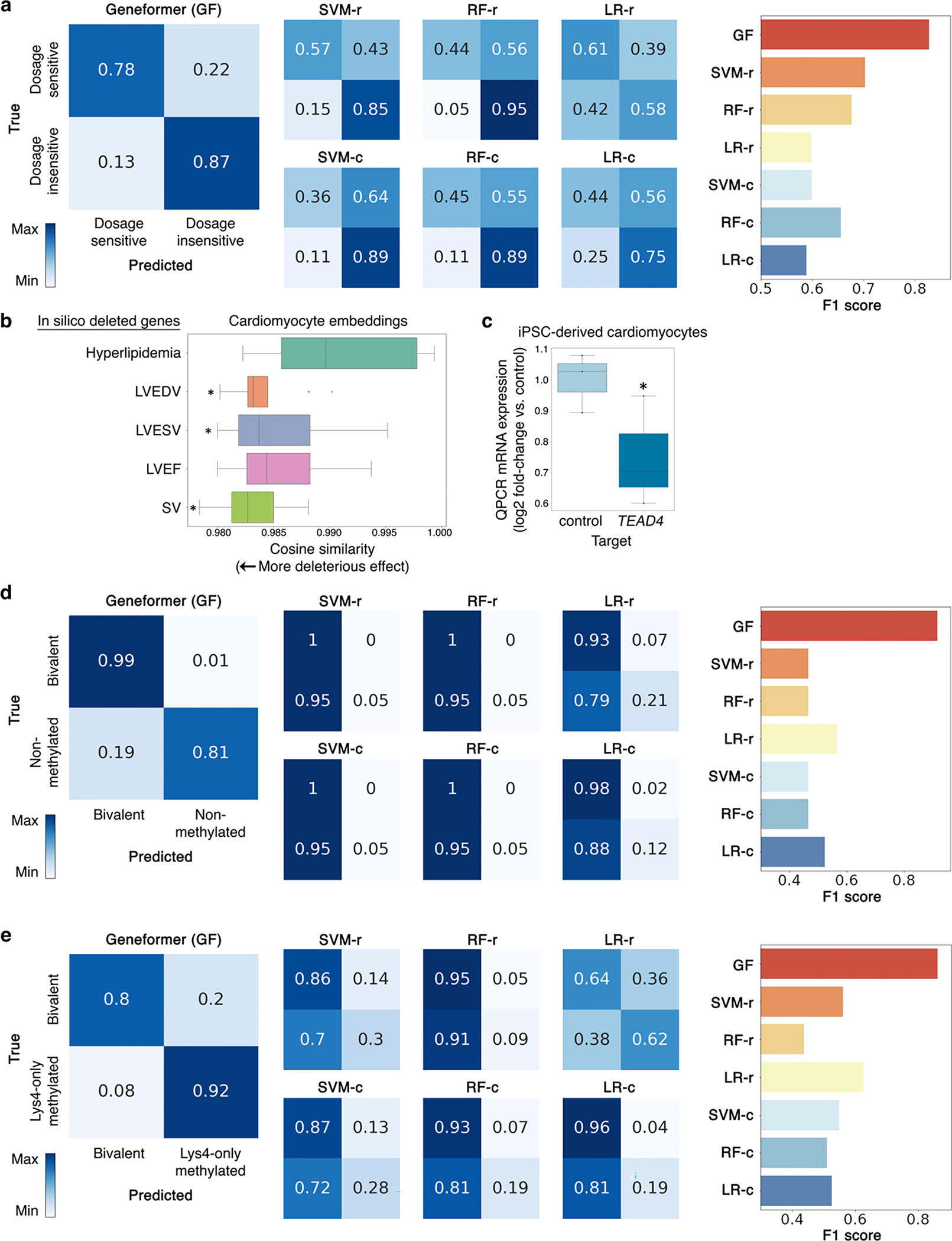
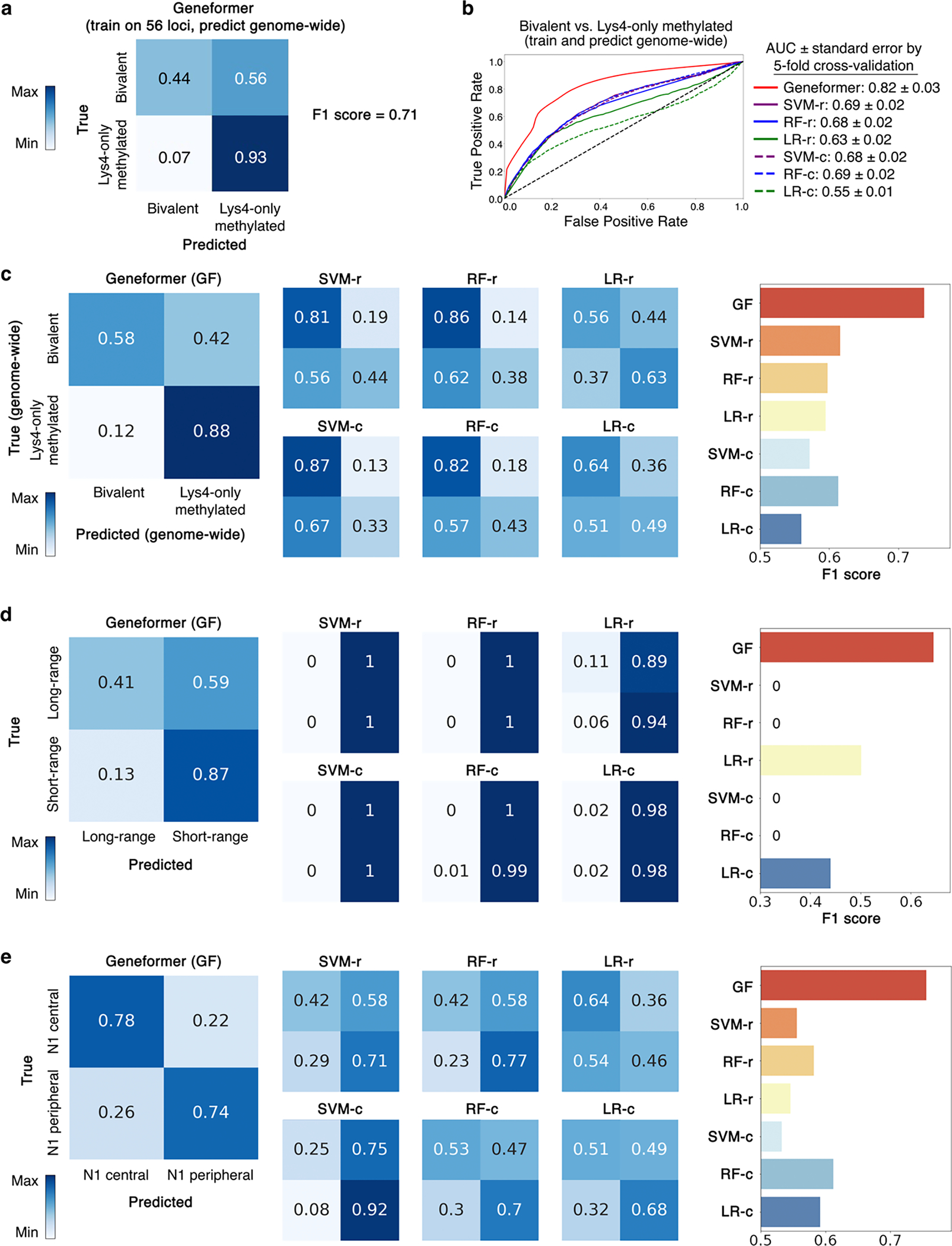
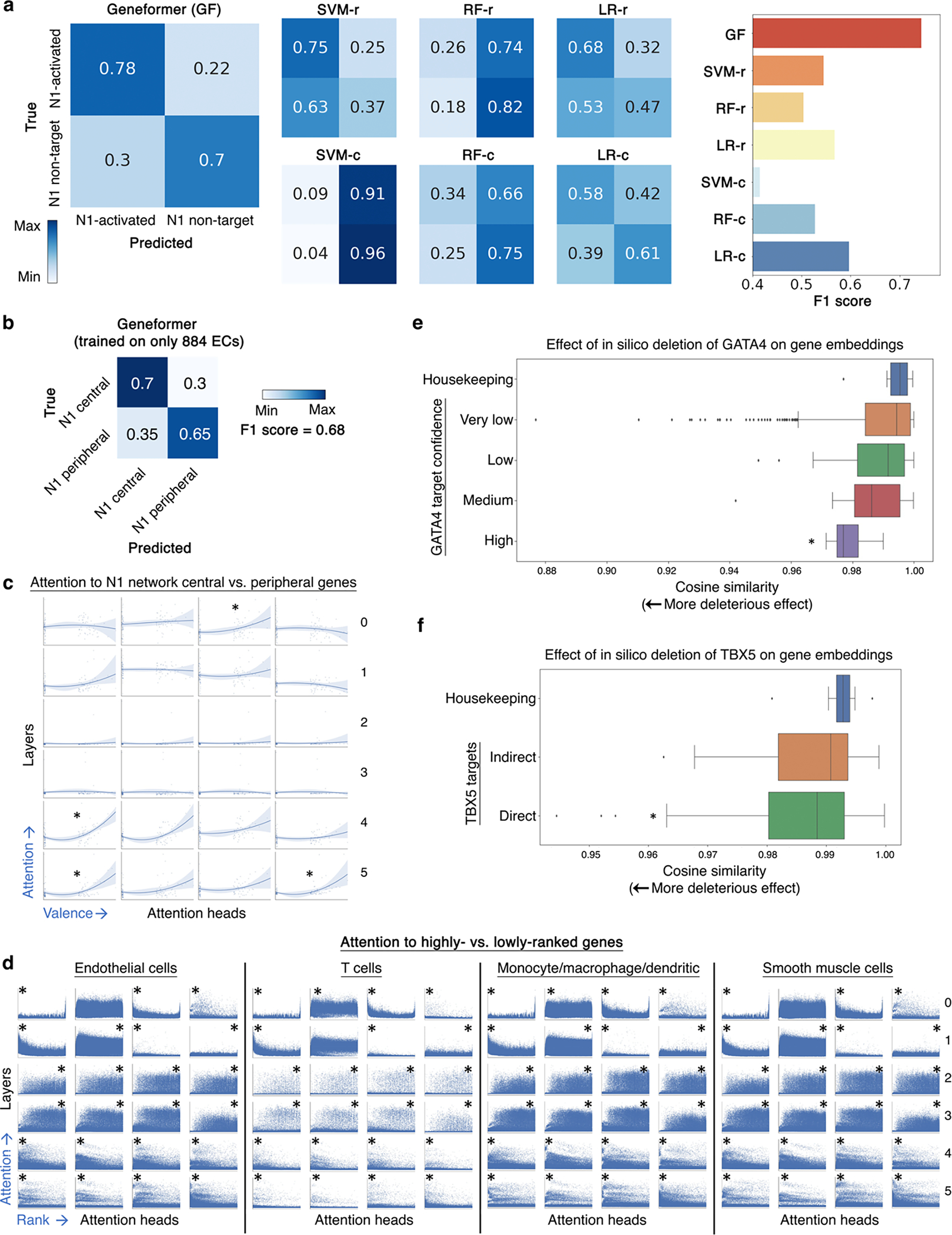
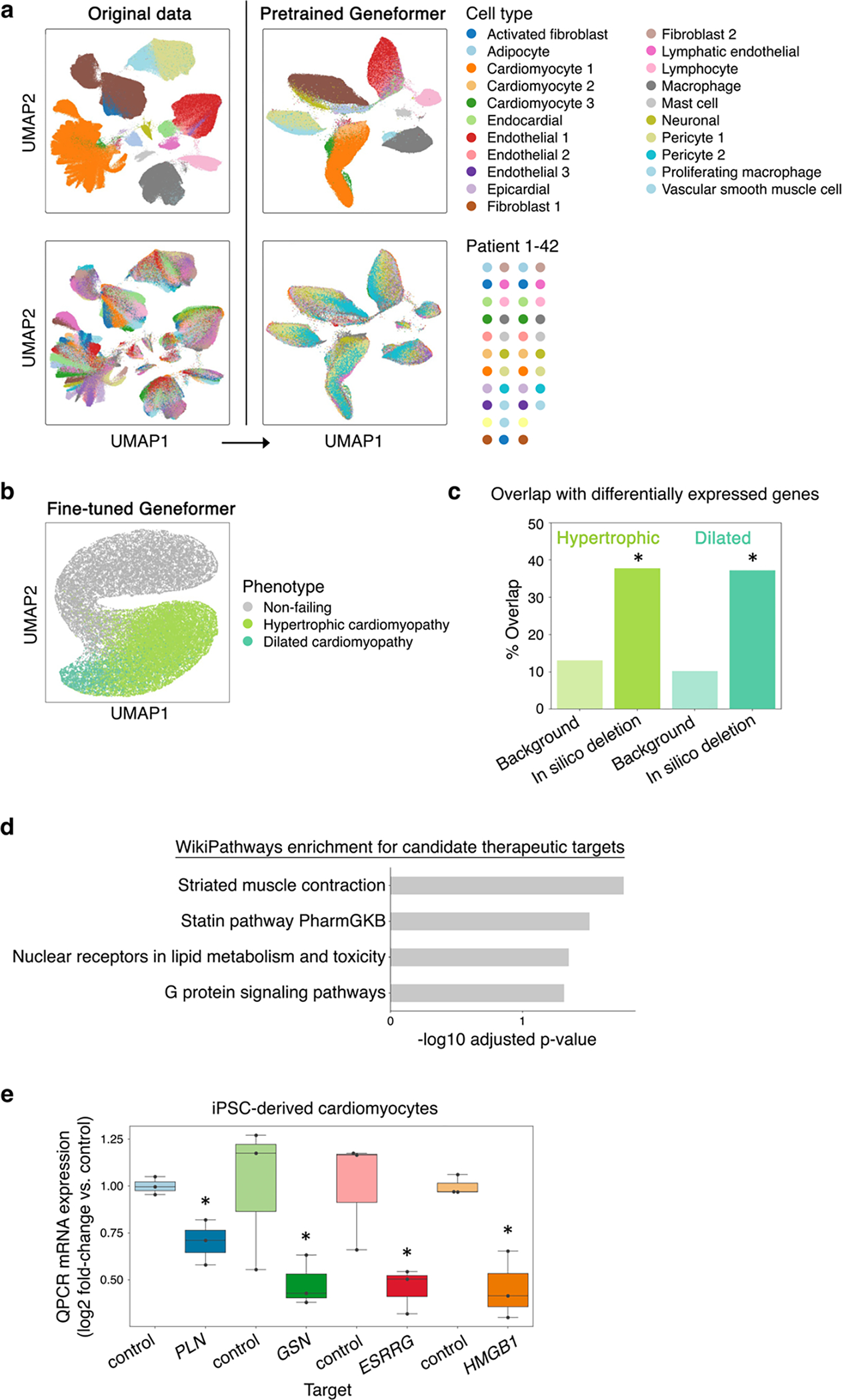
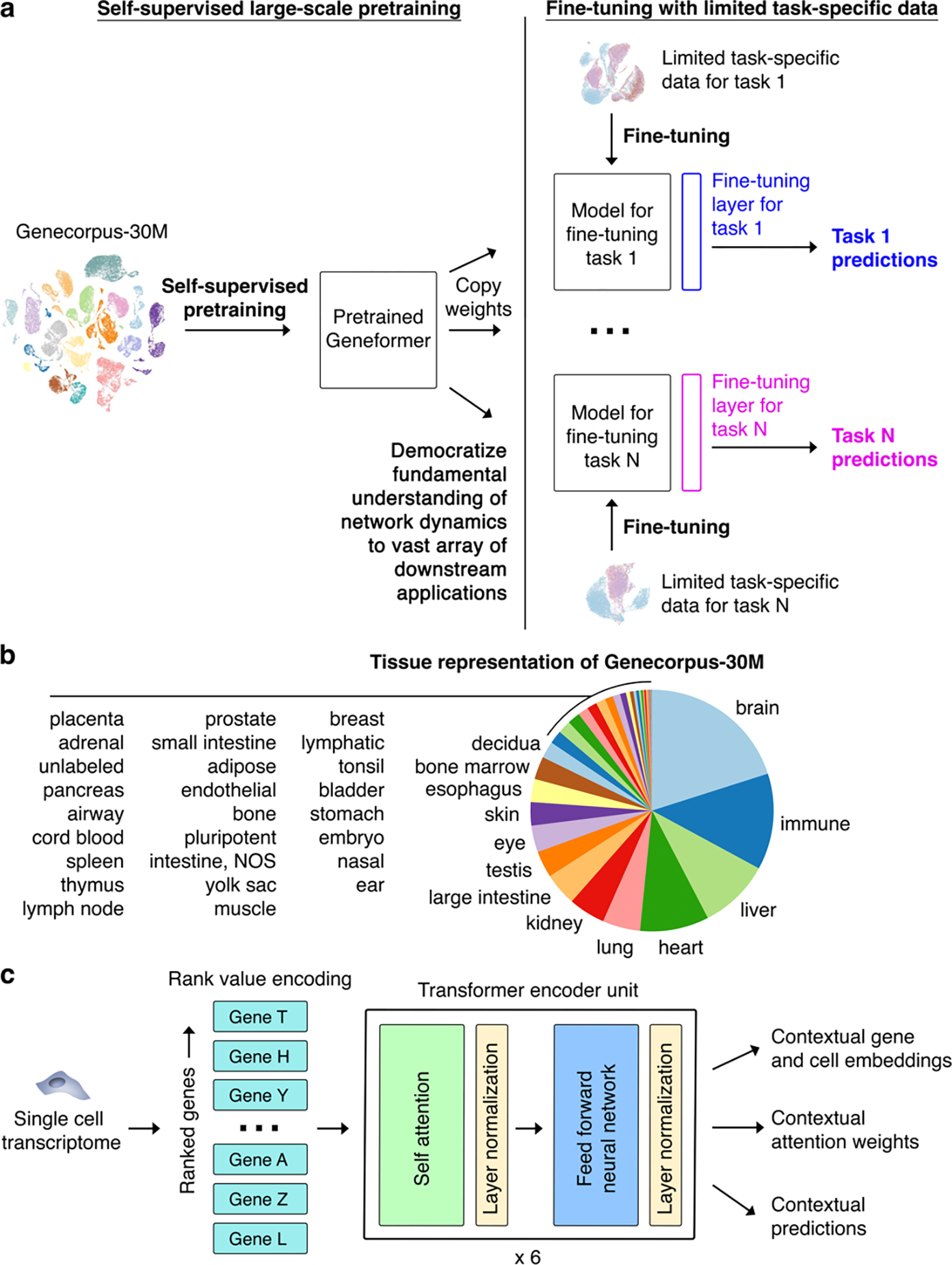
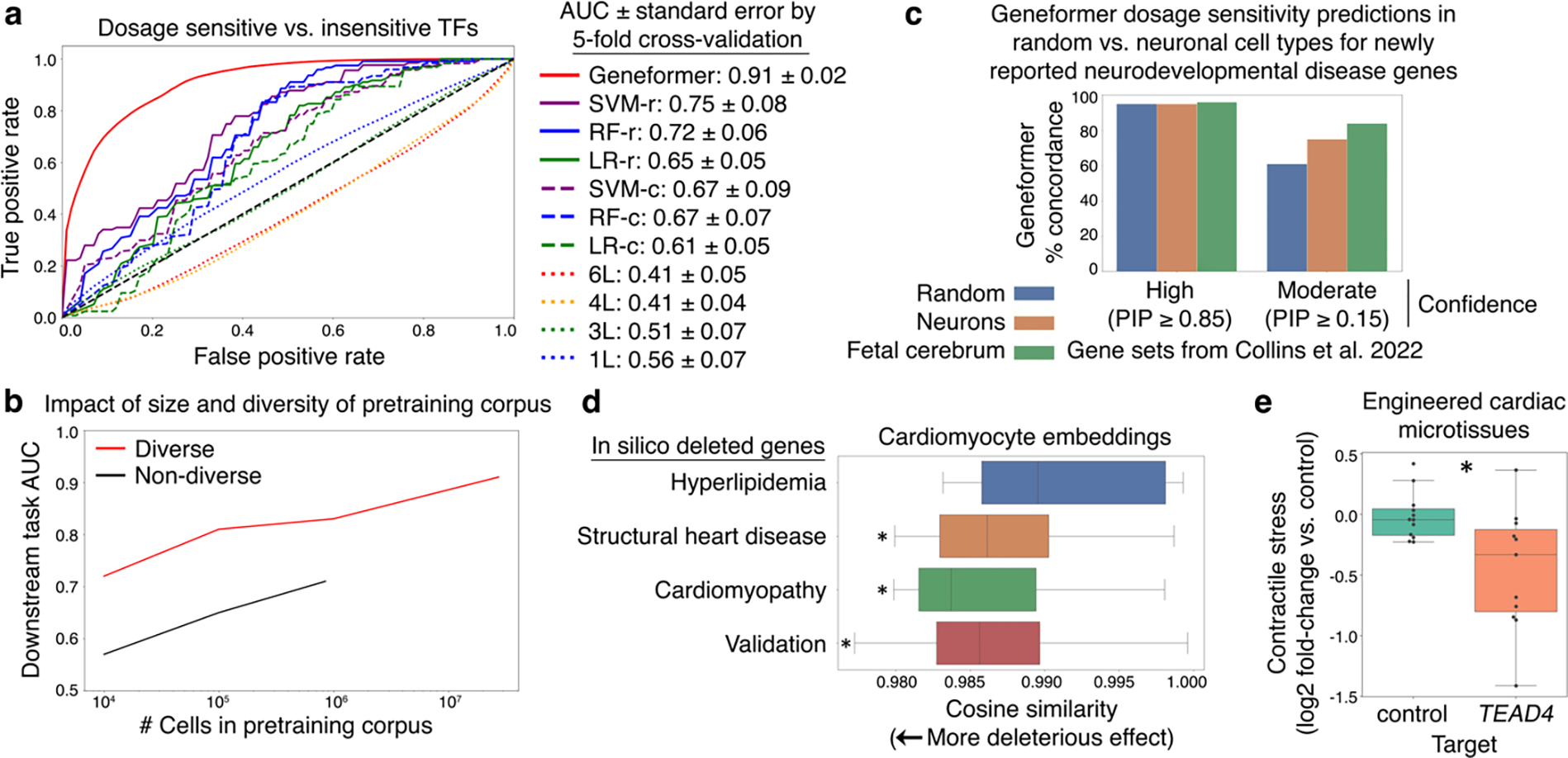
References
-
- Vaswani A Attention Is All You Need arXiv:1706.03762v5. Adv Neural Inf Process Syst 2017-Decem, (2017).
-
- Devlin J, Chang MW, Lee K & Toutanova K BERT: Pre-training of deep bidirectional transformers for language understanding. in NAACL HLT 2019 – 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies - Proceedings of the Conference vol. 1 4174–4186 (2019).
-
- He K, Zhang X, Ren S & Sun J Deep residual learning for image recognition. in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition vols 2016-December 770–778 (2016).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
