

Hierarchical Interleaved Bloom Filter: enabling ultrafast, approximate sequence queries
- PMID: 37259161
- PMCID: PMC10230713
- DOI: 10.1186/s13059-023-02971-4
Hierarchical Interleaved Bloom Filter: enabling ultrafast, approximate sequence queries
Abstract
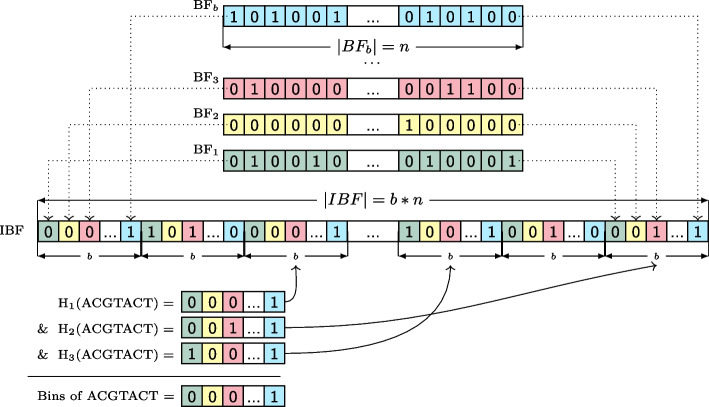
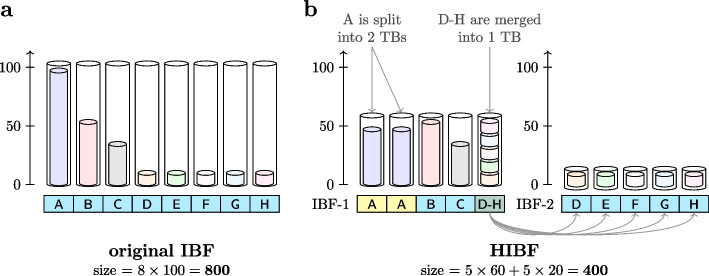
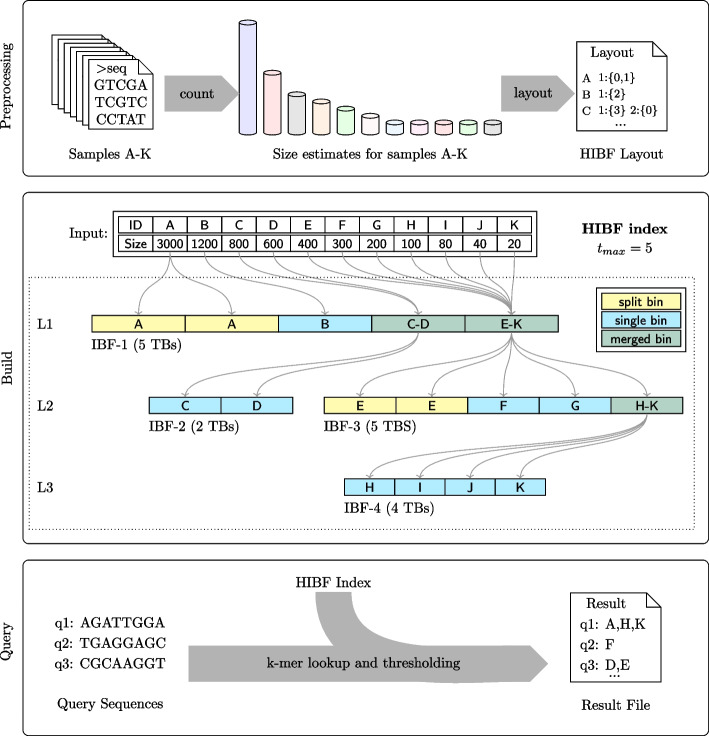
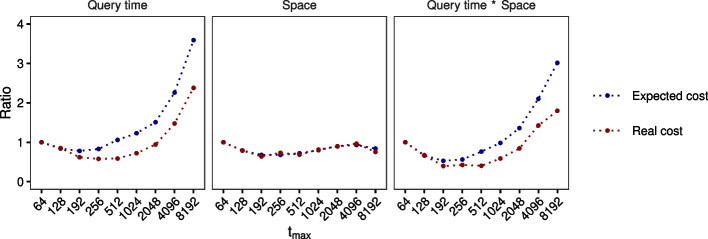
We present a novel data structure for searching sequences in large databases: the Hierarchical Interleaved Bloom Filter (HIBF). It is extremely fast and space efficient, yet so general that it could serve as the underlying engine for many applications. We show that the HIBF is superior in build time, index size, and search time while achieving a comparable or better accuracy compared to other state-of-the-art tools. The HIBF builds an index up to 211 times faster, using up to 14 times less space, and can answer approximate membership queries faster by a factor of up to 129.
Keywords: Alignment free analysis; Approximate membership query; Bloom filter; Metagenomics; Miminizer; Sequence search.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
-
- Sun C, Harris RS, Chikhi R, Medvedev P. AllSome Sequence Bloom Trees. bioRxiv. 2016;090464. 10.1101/090464. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
