

The landscape of tolerated genetic variation in humans and primates
- PMID: 37262156
- PMCID: PMC10713091
- DOI: 10.1126/science.abn8197
The landscape of tolerated genetic variation in humans and primates
Abstract
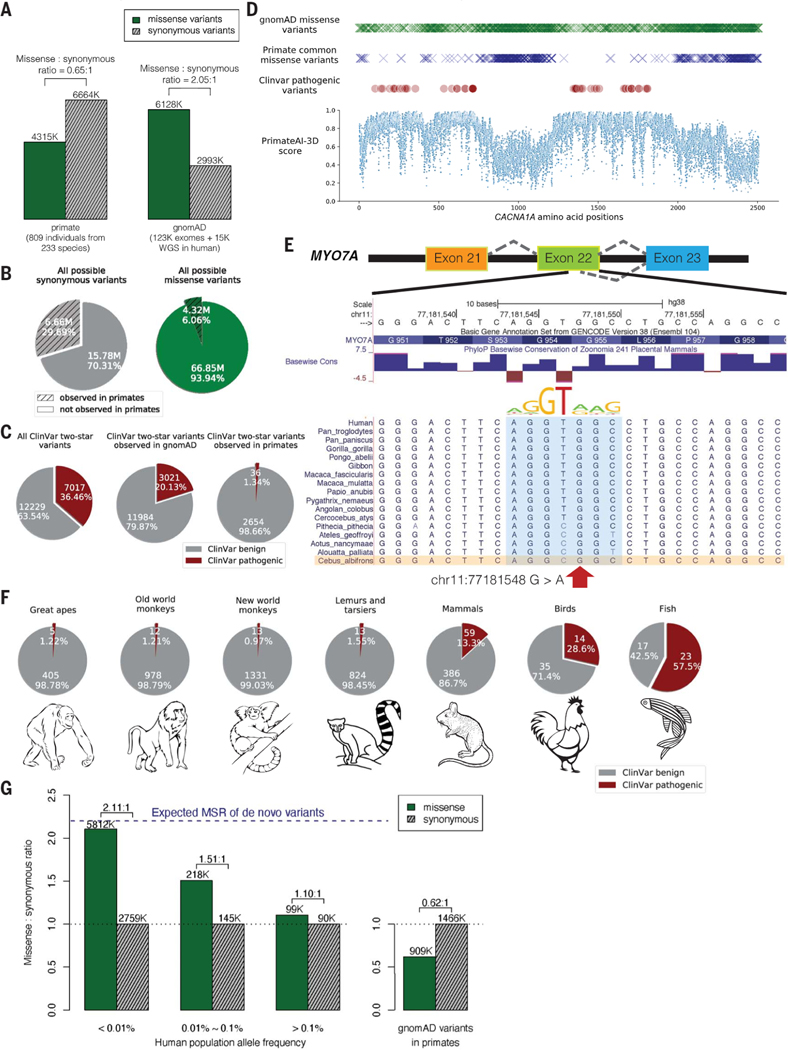
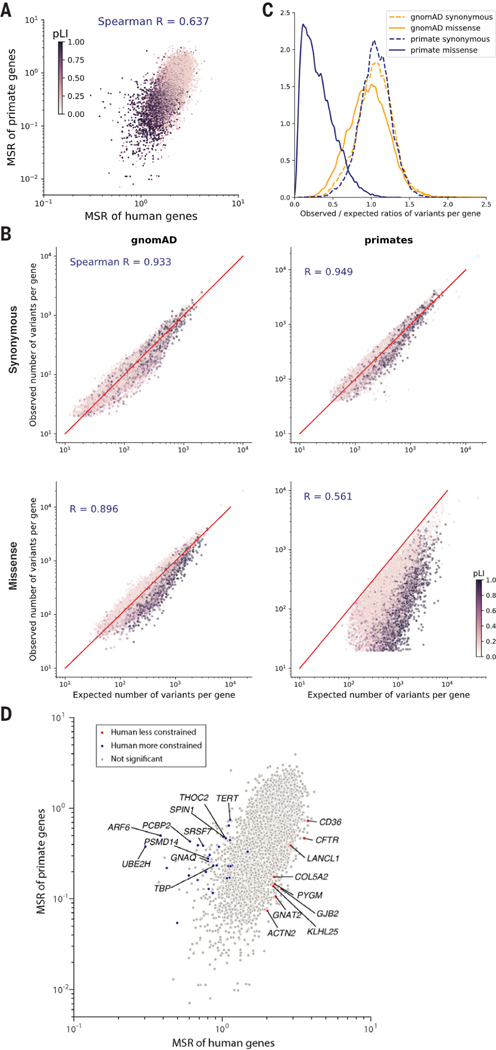
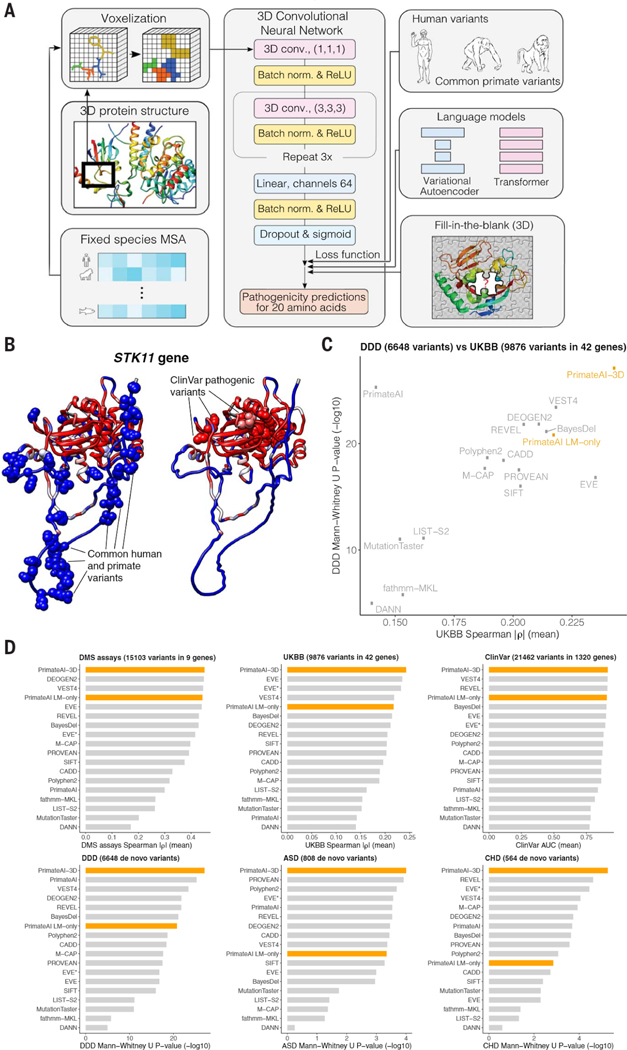
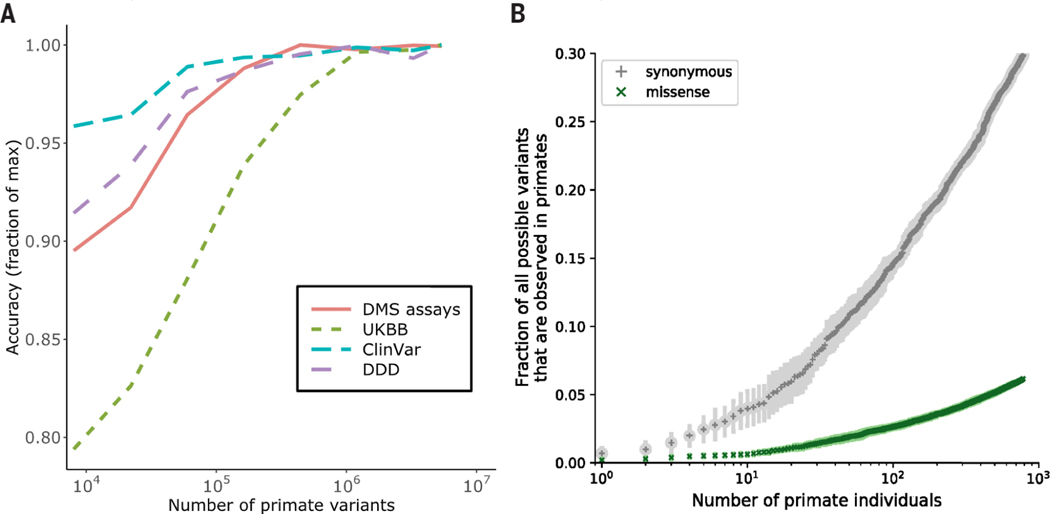
Personalized genome sequencing has revealed millions of genetic differences between individuals, but our understanding of their clinical relevance remains largely incomplete. To systematically decipher the effects of human genetic variants, we obtained whole-genome sequencing data for 809 individuals from 233 primate species and identified 4.3 million common protein-altering variants with orthologs in humans. We show that these variants can be inferred to have nondeleterious effects in humans based on their presence at high allele frequencies in other primate populations. We use this resource to classify 6% of all possible human protein-altering variants as likely benign and impute the pathogenicity of the remaining 94% of variants with deep learning, achieving state-of-the-art accuracy for diagnosing pathogenic variants in patients with genetic diseases.
Conflict of interest statement
Employees of lllumina, Inc. are indicated in the list of author affiliations. Serafim Batzoglou is currently affiliated with Seer, Inc. Heidi L. Rehm receives funding to support rare disease research and tool development from lllumina, Inc. and Microsoft, Inc. Patents related to this work are (1) title: Deep convolutional neural networks to predict variant pathogenicity using three-dimensional (3D) protein structures, filing number US 17/232,056, authors: Tobias Hamp, Kai-How Farh, Hong Gao; (2) title: Transfer learning-based use of protein contact maps for variant pathogenicity prediction, filing No.: US 17/876,481, authors: Chen Chen, Hong Gao, Laksshman Sundaram, Kai-How Farh; (3) title: Multichannel protein voxelization to predict variant pathogenicity using deep convolutional neural networks, filing number US 17/703,935, authors: Tobias Hamp, Kai-How Farh, Hong Gao;(4) title: Transformer language model for variant pathogenicity, filing number US 17/975,536 and US 17/975,547, authors: Jeffrey Ede, Tobias Hamp, Anastasia Dietrich, Yibing Wu, Kai-How Farh. (5) title: Identifying genes with differential selective constraint between humans and nonhuman primates, filing number US 63/294,820, authors: H. G., J. G. Schraiber, K.-H. Farh.
Figures
Update of
-
The landscape of tolerated genetic variation in humans and primates.bioRxiv [Preprint]. 2023 May 2:2023.05.01.538953. doi: 10.1101/2023.05.01.538953. bioRxiv. 2023. Update in: Science. 2023 Jun 2;380(6648):eabn8153. doi: 10.1126/science.abn8197. PMID: 37205491 Free PMC article. Updated. Preprint.
Comment in
-
Improved pathogenicity prediction using primate genomics.Nat Genet. 2023 Jul;55(7):1082. doi: 10.1038/s41588-023-01455-2. Nat Genet. 2023. PMID: 37438535 No abstract available.
References
-
- Nussbaum RL, Rehm HL; ClinGen, ClinGen and Genetic Testing. N. Engl. J. Med 373,1379 (2015). pmid: 26430707 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
