

An ERK5-NRF2 Axis Mediates Senescence-Associated Stemness and Atherosclerosis
- PMID: 37264926
- PMCID: PMC10357365
- DOI: 10.1161/CIRCRESAHA.122.322017
An ERK5-NRF2 Axis Mediates Senescence-Associated Stemness and Atherosclerosis
Abstract
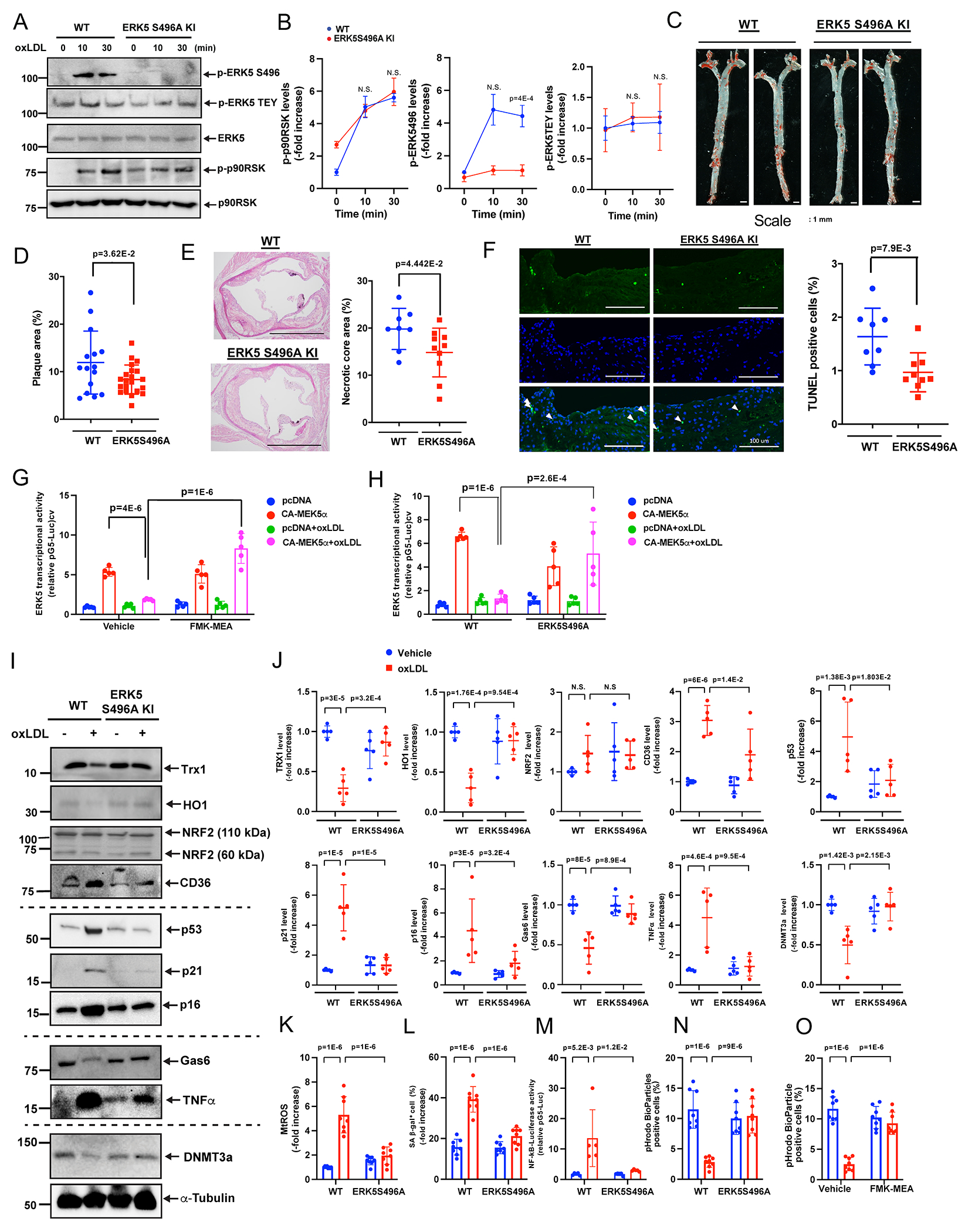
Background: ERK5 (extracellular signal-regulated kinase 5) is a dual kinase transcription factor containing an N-terminal kinase domain and a C-terminal transcriptional activation domain. Many ERK5 kinase inhibitors have been developed and tested to treat cancer and inflammatory diseases. However, recent data have raised questions about the role of the catalytic activity of ERK5 in proliferation and inflammation. We aimed to investigate how ERK5 reprograms myeloid cells to the proinflammatory senescent phenotype, subsequently leading to atherosclerosis.
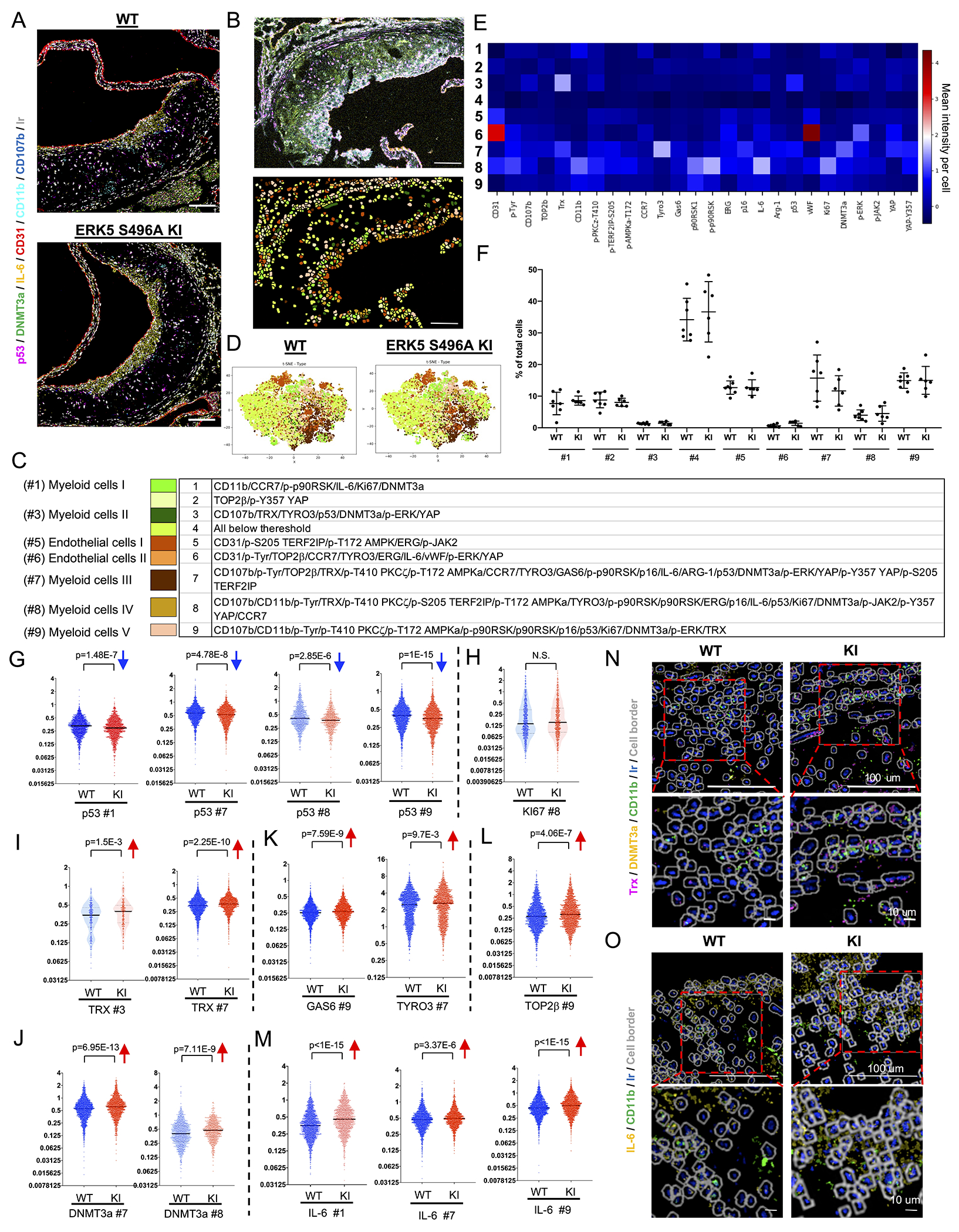
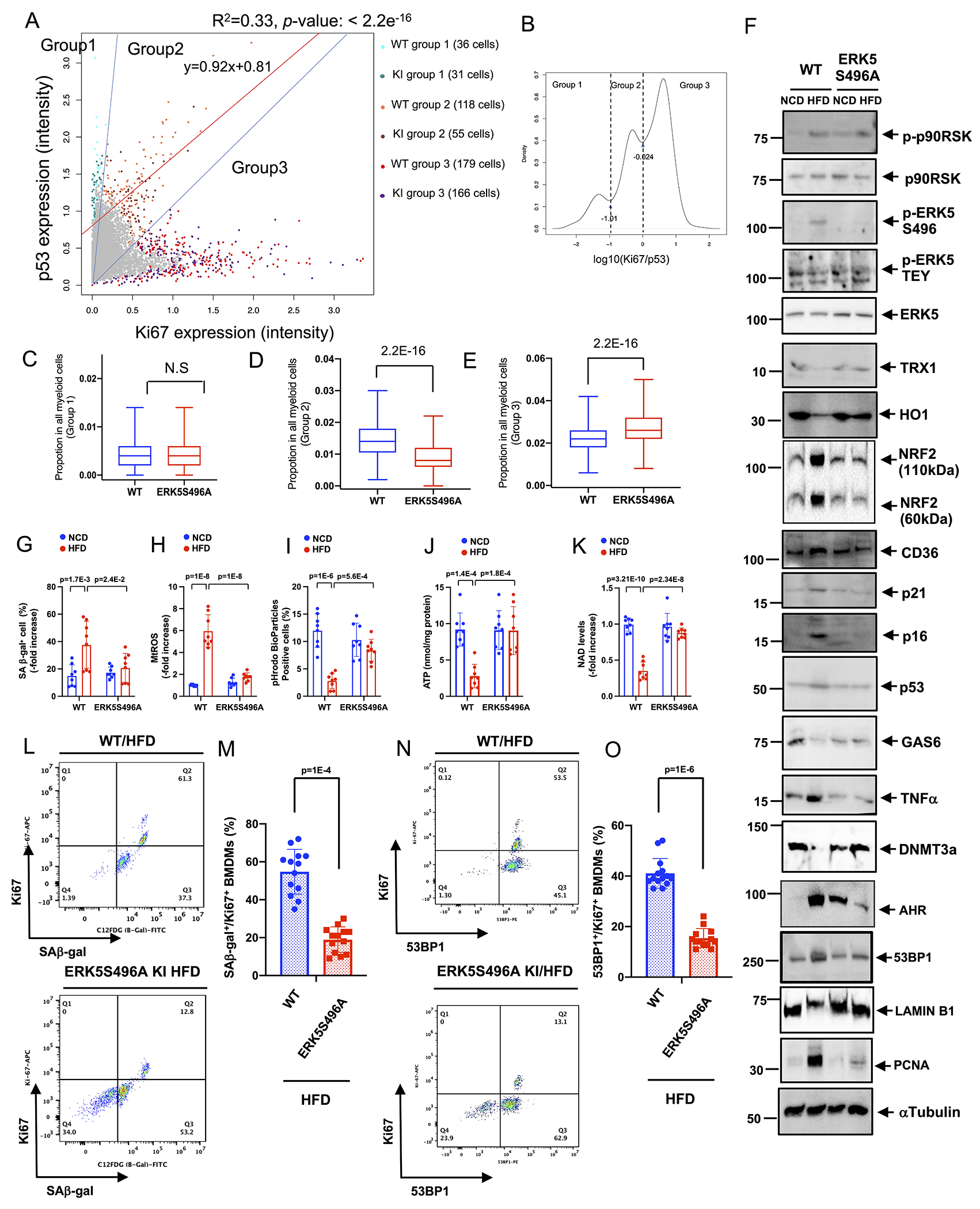
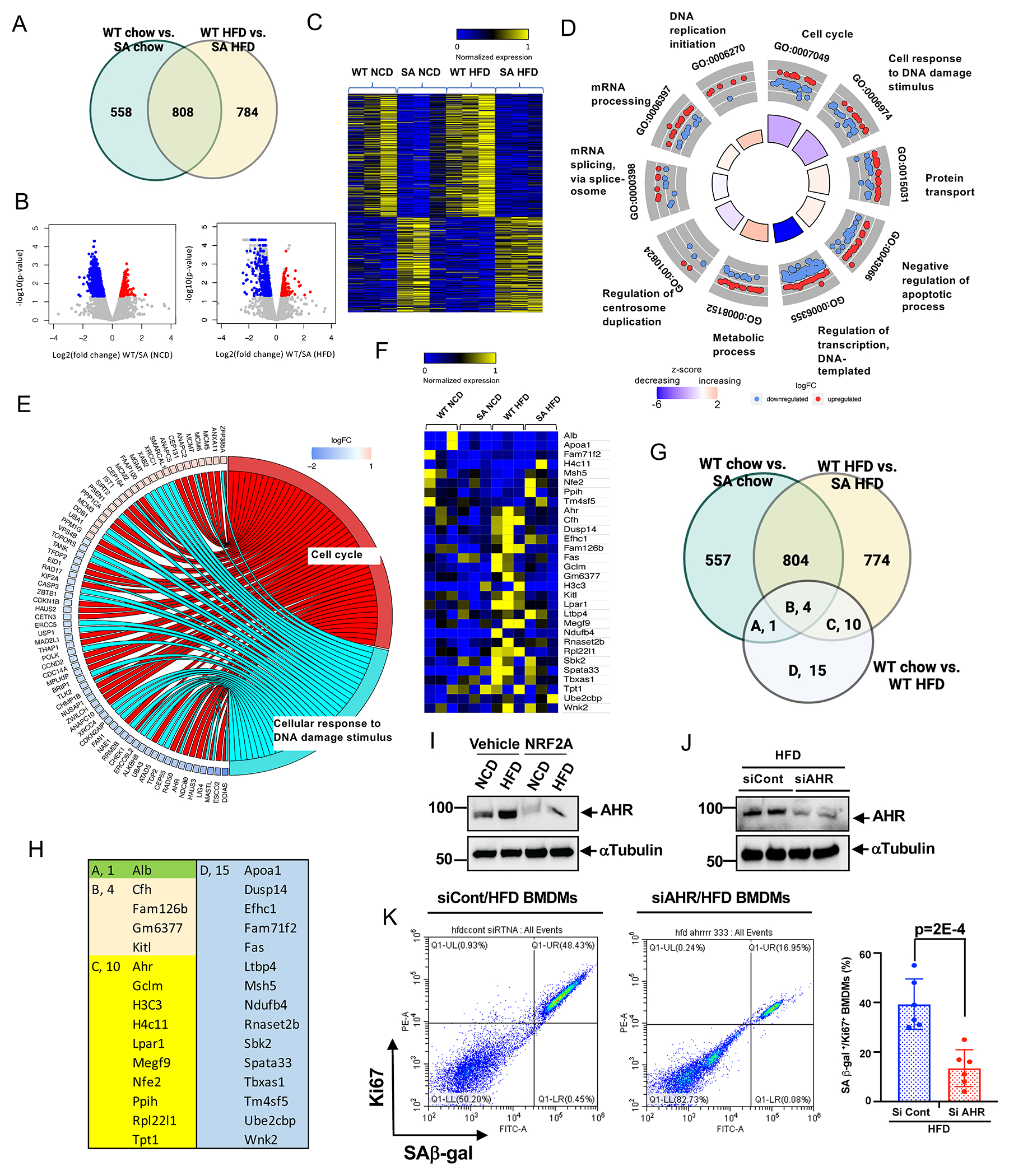
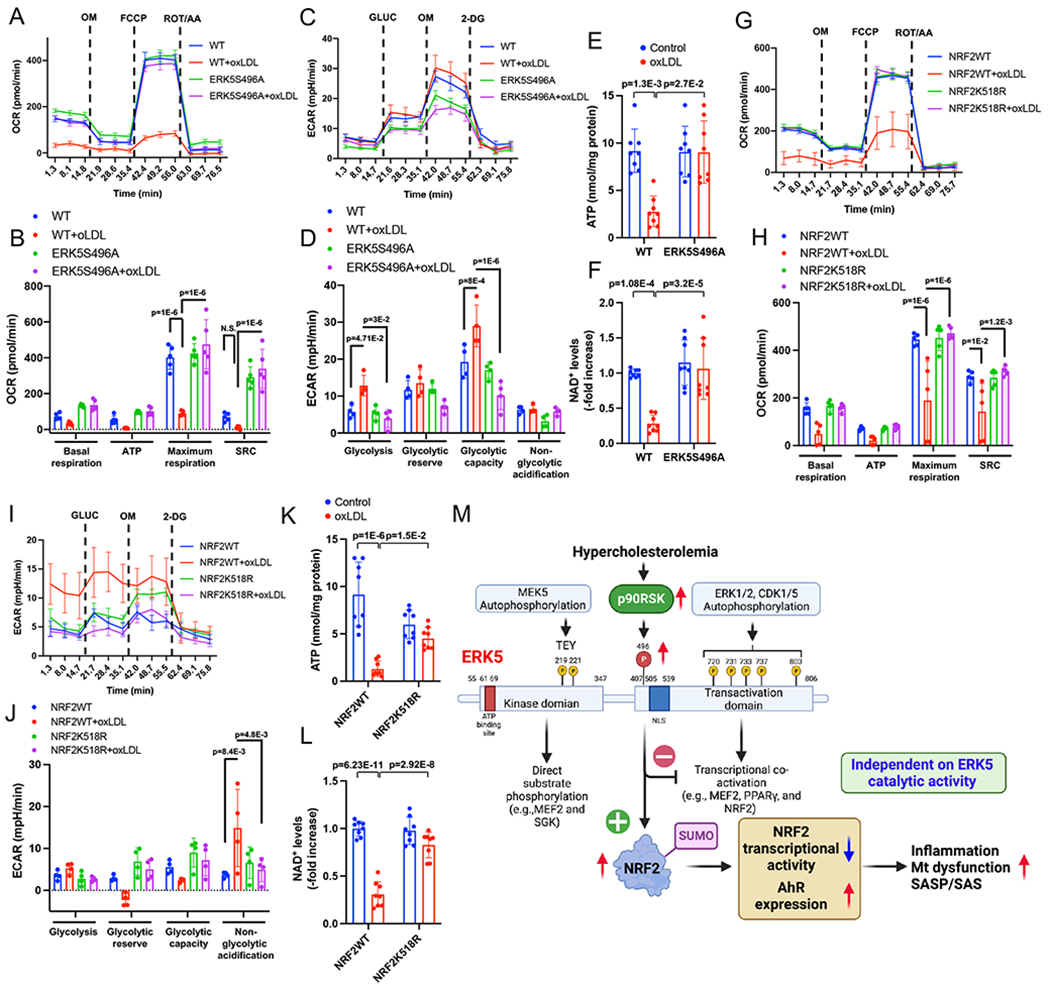
Methods: A ERK5 S496A (dephosphorylation mimic) knock in (KI) mouse model was generated using CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeat-associated 9), and atherosclerosis was characterized by hypercholesterolemia induction. The plaque phenotyping in homozygous ERK5 S496A KI and wild type (WT) mice was studied using imaging mass cytometry. Bone marrow-derived macrophages were isolated from hypercholesterolemic mice and characterized using RNA sequencing and functional in vitro approaches, including senescence, mitochondria reactive oxygen species, and inflammation assays, as well as by metabolic extracellular flux analysis.
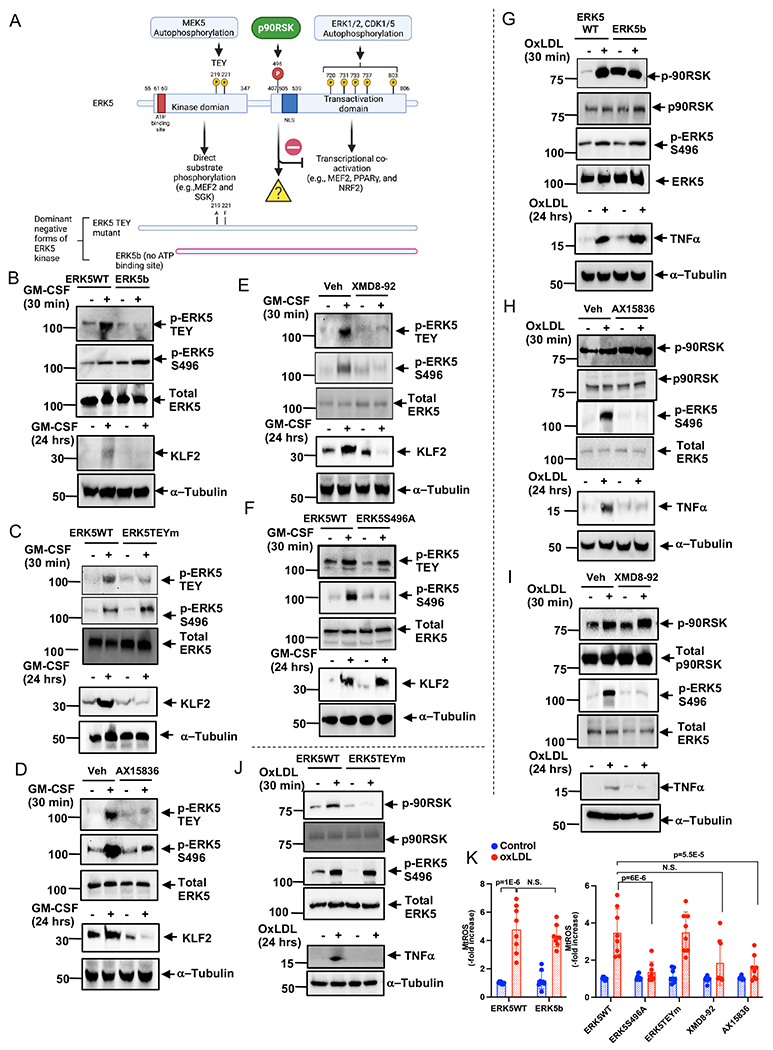
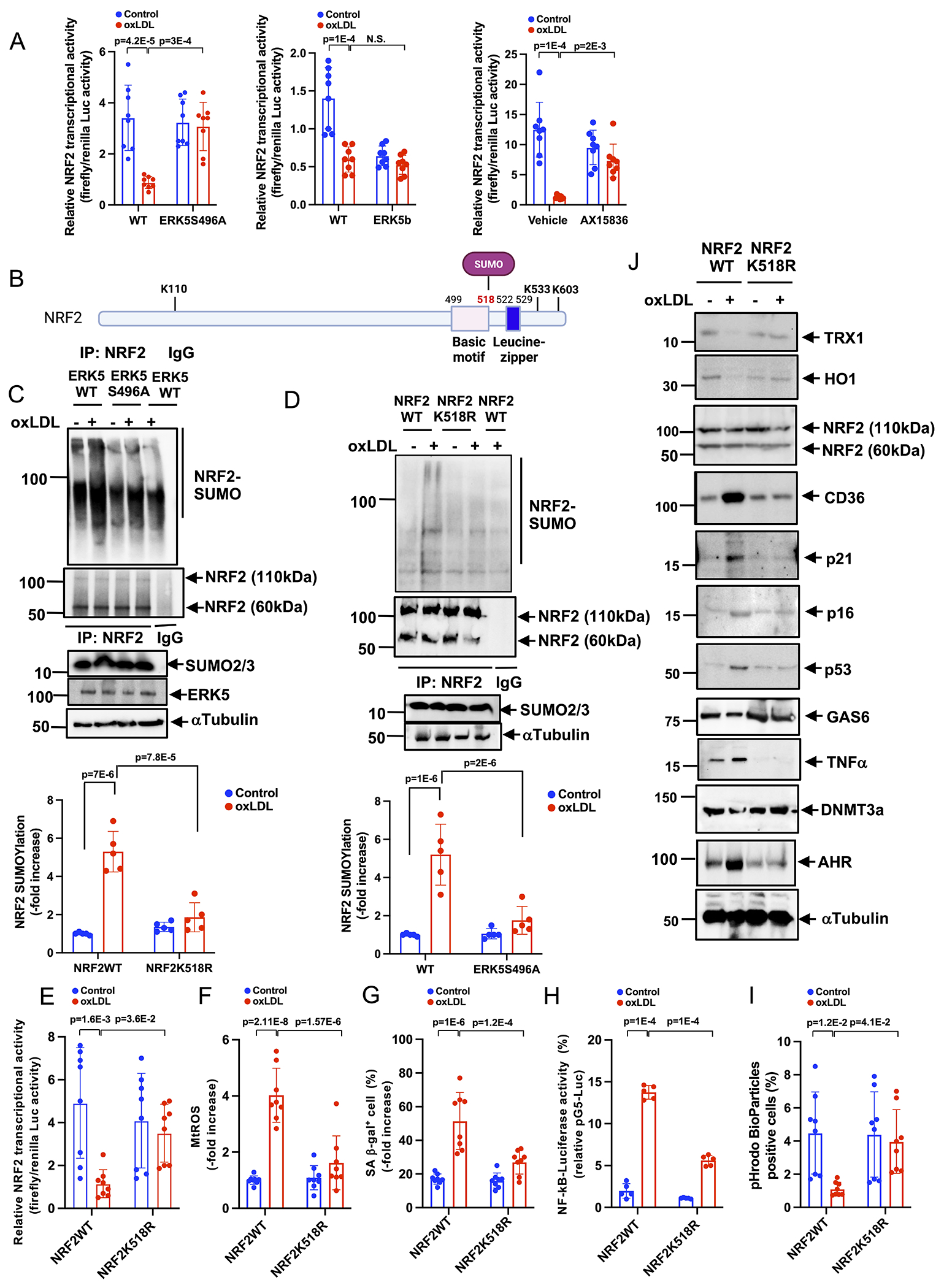
Results: We show that atherosclerosis was inhibited in ERK5 S496A KI mice. Furthermore, ERK5 S496 phosphorylation mediates both senescence-associated secretory phenotype and senescence-associated stemness by upregulating AHR (aryl hydrocarbon receptor) in plaque and bone marrow-derived macrophages isolated from hypercholesterolemic mice. We also discovered that ERK5 S496 phosphorylation could induce NRF2 (NFE2-related factor 2) SUMOylation at a novel K518 site to inhibit NRF2 transcriptional activity without altering ERK5 catalytic activity and mediates oxidized LDL (low-density lipoprotein)-induced senescence-associated secretory phenotype. Specific ERK5 kinase inhibitors (AX15836 and XMD8-92) also inhibited ERK5 S496 phosphorylation, suggesting the involvement of ERK5 S496 phosphorylation in the anti-inflammatory effects of these ERK5 kinase inhibitors.
Conclusions: We discovered a novel mechanism by which the macrophage ERK5-NRF2 axis develops a unique senescence-associated secretory phenotype/stemness phenotype by upregulating AHR to engender atherogenesis. The finding of senescence-associated stemness phenotype provides a molecular explanation to resolve the paradox of senescence in proliferative plaque by permitting myeloid cells to escape the senescence-induced cell cycle arrest during atherosclerosis formation.
Keywords: NF-E2-related factor 2 (NRF2); antioxidants; atherosclerosis; mitogen-activated protein kinase 7 (ERK5); receptors, aryl hydrocarbon (AHR); senescence-associated secretory phenotype; sumoylation.
Conflict of interest statement
Figures
References
-
- Singh MV, Kotla S, Le NT, Ae Ko K, Heo KS, Wang Y, Fujii Y, Thi Vu H, McBeath E, Thomas TN, et al. Senescent Phenotype Induced by p90RSK-NRF2 Signaling Sensitizes Monocytes and Macrophages to Oxidative Stress in HIV-Positive Individuals. Circulation. 2019;139:1199–1216. doi: 10.1161/CIRCULATIONAHA.118.036232 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
