

pyCHARMM: Embedding CHARMM Functionality in a Python Framework
- PMID: 37267404
- PMCID: PMC10504603
- DOI: 10.1021/acs.jctc.3c00364
pyCHARMM: Embedding CHARMM Functionality in a Python Framework
Abstract
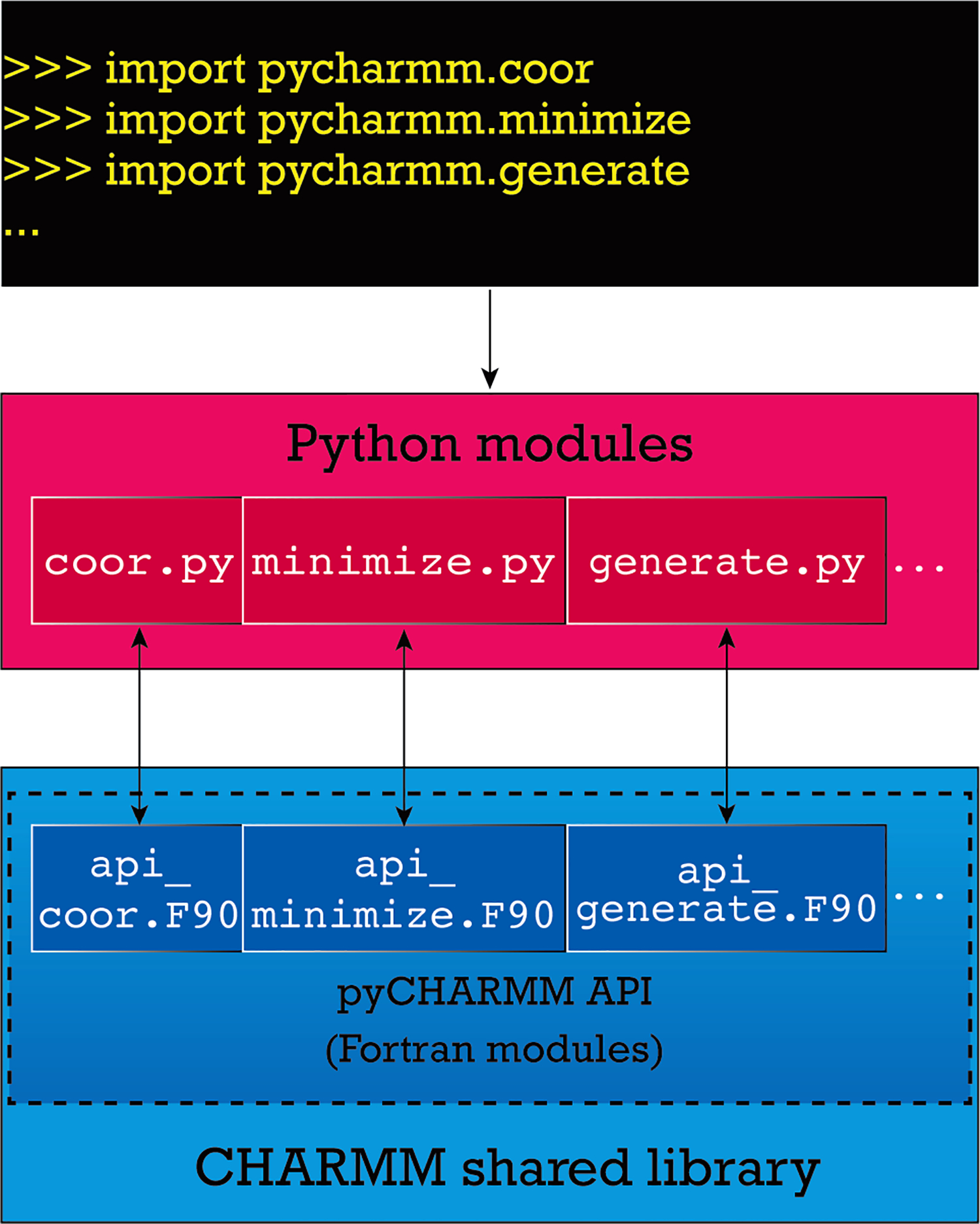
CHARMM is rich in methodology and functionality as one of the first programs addressing problems of molecular dynamics and modeling of biological macromolecules and their partners, e.g., small molecule ligands. When combined with the highly developed CHARMM parameters for proteins, nucleic acids, small molecules, lipids, sugars, and other biologically relevant building blocks, and the versatile CHARMM scripting language, CHARMM has been a trendsetting platform for modeling studies of biological macromolecules. To further enhance the utility of accessing and using CHARMM functionality in increasingly complex workflows associated with modeling biological systems, we introduce pyCHARMM, Python bindings, functions, and modules to complement and extend the extensive set of modeling tools and methods already available in CHARMM. These include access to CHARMM function-generated variables associated with the system (psf), coordinates, velocities and forces, atom selection variables, and force field related parameters. The ability to augment CHARMM forces and energies with energy terms or methods derived from machine learning or other sources, written in Python, CUDA, or OpenCL and expressed as Python callable routines is introduced together with analogous functions callable during dynamics calculations. Integration of Python-based graphical engines for visualization of simulation models and results is also accessible. Loosely coupled parallelism is available for workflows such as free energy calculations, using MBAR/TI approaches or high-throughput multisite λ-dynamics (MSλD) free energy methods, string path optimization calculations, replica exchange, and molecular docking with a new Python-based CDOCKER module. CHARMM accelerated platform kernels through the CHARMM/OpenMM API, CHARMM/DOMDEC, and CHARMM/BLaDE API are also readily integrated into this Python framework. We anticipate that pyCHARMM will be a robust platform for the development of comprehensive and complex workflows utilizing Python and its extensive functionality as well as an optimal platform for users to learn molecular modeling methods and practices within a Python-friendly environment such as Jupyter Notebooks.
Conflict of interest statement
Notes
The authors declare no competing financial interest.
Figures





Similar articles
-
CHARMM-GUI 10 years for biomolecular modeling and simulation.J Comput Chem. 2017 Jun 5;38(15):1114-1124. doi: 10.1002/jcc.24660. Epub 2016 Nov 14. J Comput Chem. 2017. PMID: 27862047 Free PMC article. Review.
-
BLaDE: A Basic Lambda Dynamics Engine for GPU-Accelerated Molecular Dynamics Free Energy Calculations.J Chem Theory Comput. 2021 Nov 9;17(11):6799-6807. doi: 10.1021/acs.jctc.1c00833. Epub 2021 Oct 28. J Chem Theory Comput. 2021. PMID: 34709046 Free PMC article.
-
TopoGromacs: Automated Topology Conversion from CHARMM to GROMACS within VMD.J Chem Inf Model. 2016 Jun 27;56(6):1112-6. doi: 10.1021/acs.jcim.6b00103. Epub 2016 Jun 1. J Chem Inf Model. 2016. PMID: 27196035 Free PMC article.
-
CHARMM: the biomolecular simulation program.J Comput Chem. 2009 Jul 30;30(10):1545-614. doi: 10.1002/jcc.21287. J Comput Chem. 2009. PMID: 19444816 Free PMC article. Review.
-
CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules.J Comput Chem. 2017 Jun 5;38(21):1879-1886. doi: 10.1002/jcc.24829. Epub 2017 May 11. J Comput Chem. 2017. PMID: 28497616 Free PMC article.
Cited by
-
Simultaneous Construction of Free Energy Surfaces via Multisite λ Dynamics and Umbrella Sampling.J Chem Theory Comput. 2025 Sep 9;21(17):8320-8329. doi: 10.1021/acs.jctc.5c00807. Epub 2025 Aug 20. J Chem Theory Comput. 2025. PMID: 40834339 Free PMC article.
-
Enhancing Empirical Energy Functions Using Physics- and Machine Learning-Based Extensions: Structure, Dynamics and Spectroscopy of Modified Benzenes.J Comput Chem. 2025 Aug 5;46(21):e70162. doi: 10.1002/jcc.70162. J Comput Chem. 2025. PMID: 40751928 Free PMC article.
-
Structural Modeling of Cytokine-Receptor-JAK2 Signaling Complexes Using AlphaFold Multimer.J Chem Inf Model. 2023 Sep 25;63(18):5874-5895. doi: 10.1021/acs.jcim.3c00926. Epub 2023 Sep 11. J Chem Inf Model. 2023. PMID: 37694948 Free PMC article.
-
Guiding discovery of protein sequence-structure-function modeling.Bioinformatics. 2024 Jan 2;40(1):btae002. doi: 10.1093/bioinformatics/btae002. Bioinformatics. 2024. PMID: 38195719 Free PMC article.
-
Ancestral Sequence Reconstruction to Enable Biocatalytic Synthesis of Azaphilones.J Am Chem Soc. 2024 Nov 6;146(44):30194-30203. doi: 10.1021/jacs.4c08761. Epub 2024 Oct 23. J Am Chem Soc. 2024. PMID: 39441831 Free PMC article.
References
-
- Macuglia D; Roux B; Ciccotti G The emergence of protein dynamics simulations: how computational statistical mechanics met biochemistry. Eur. Phys. J. H 2022, 47, 13. DOI: 10.1140/epjh/s13129-022-00043-y. - DOI
-
- Hockney RW; Eastwood JW Computer Simulation Using Particles; McGraw-Hill, 1981.
-
- McCammon JA; Harvey S Dynamics of Proteins and Nucleic Acids; Cambridge University Press, 1987.
-
- Brooks CL III; Karplus M; Pettitt BM Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics; John Wiley & Sons, 1988.
-
- Allen MP; Tildesley DJ Computer Simulation of Liquids; Oxford University Press, 1989.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources