

Transcriptional space-time mapping identifies concerted immune and stromal cell patterns and gene programs in wound healing and cancer
- PMID: 37267918
- PMCID: PMC10843988
- DOI: 10.1016/j.stem.2023.05.001
Transcriptional space-time mapping identifies concerted immune and stromal cell patterns and gene programs in wound healing and cancer
Abstract
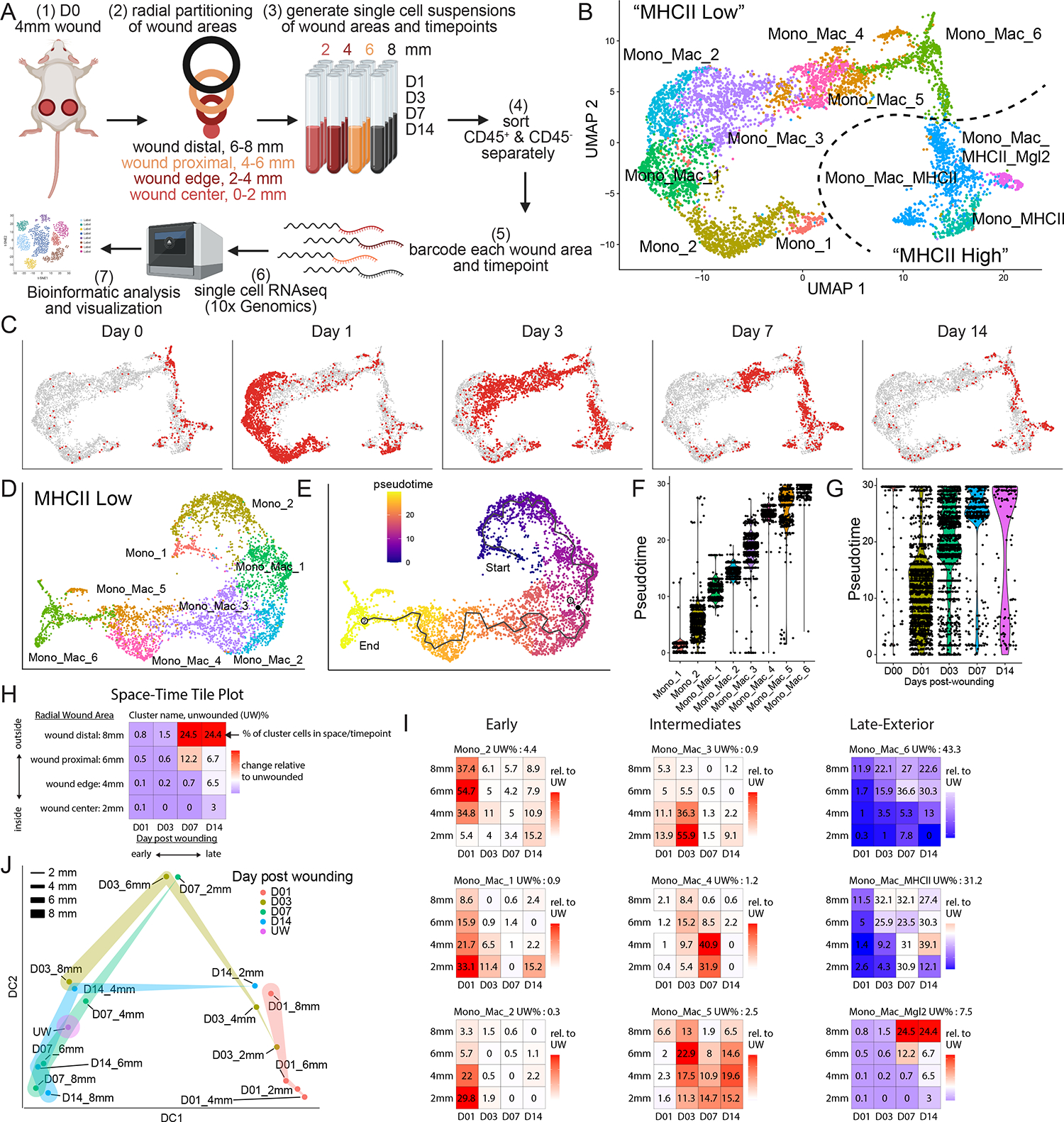
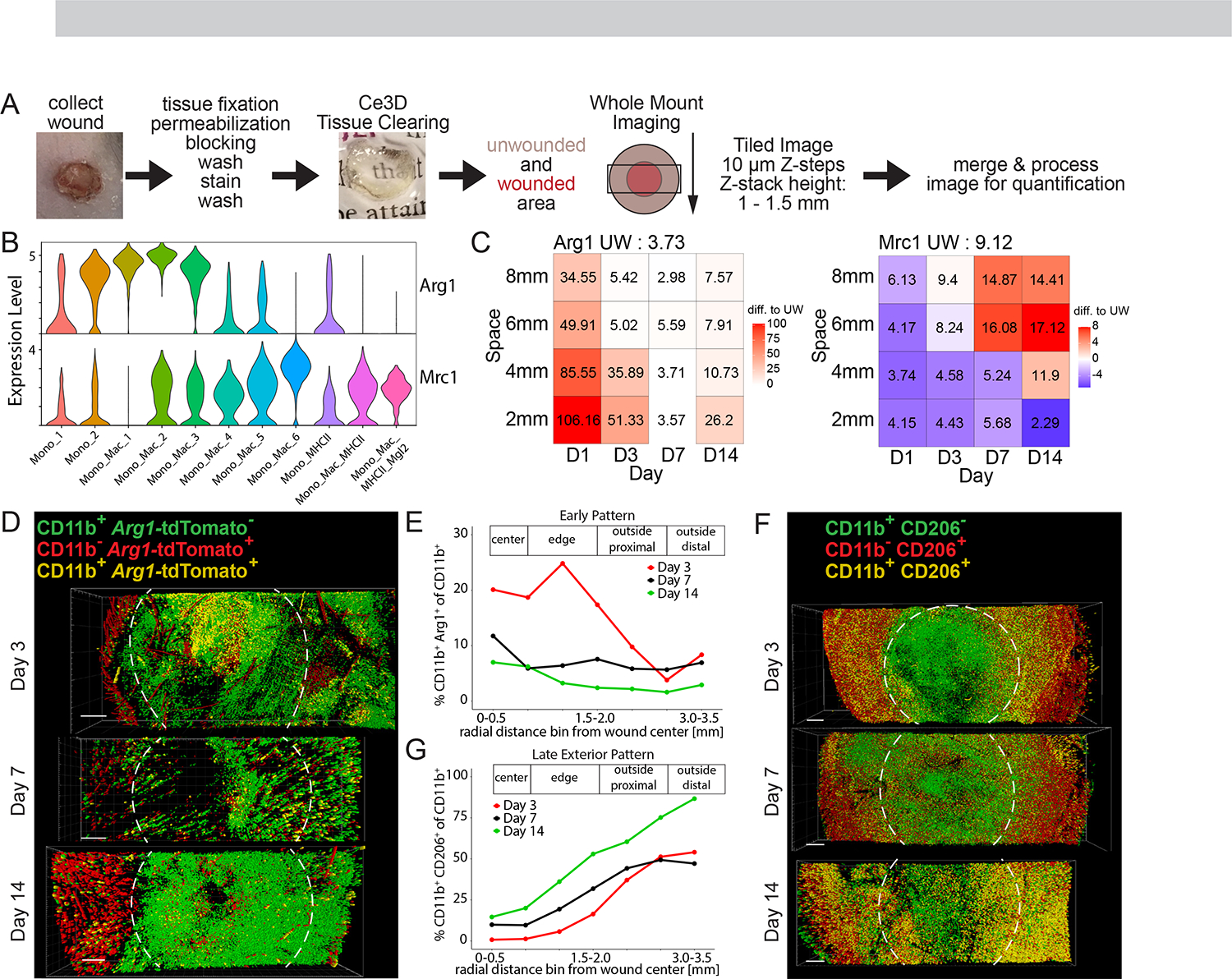
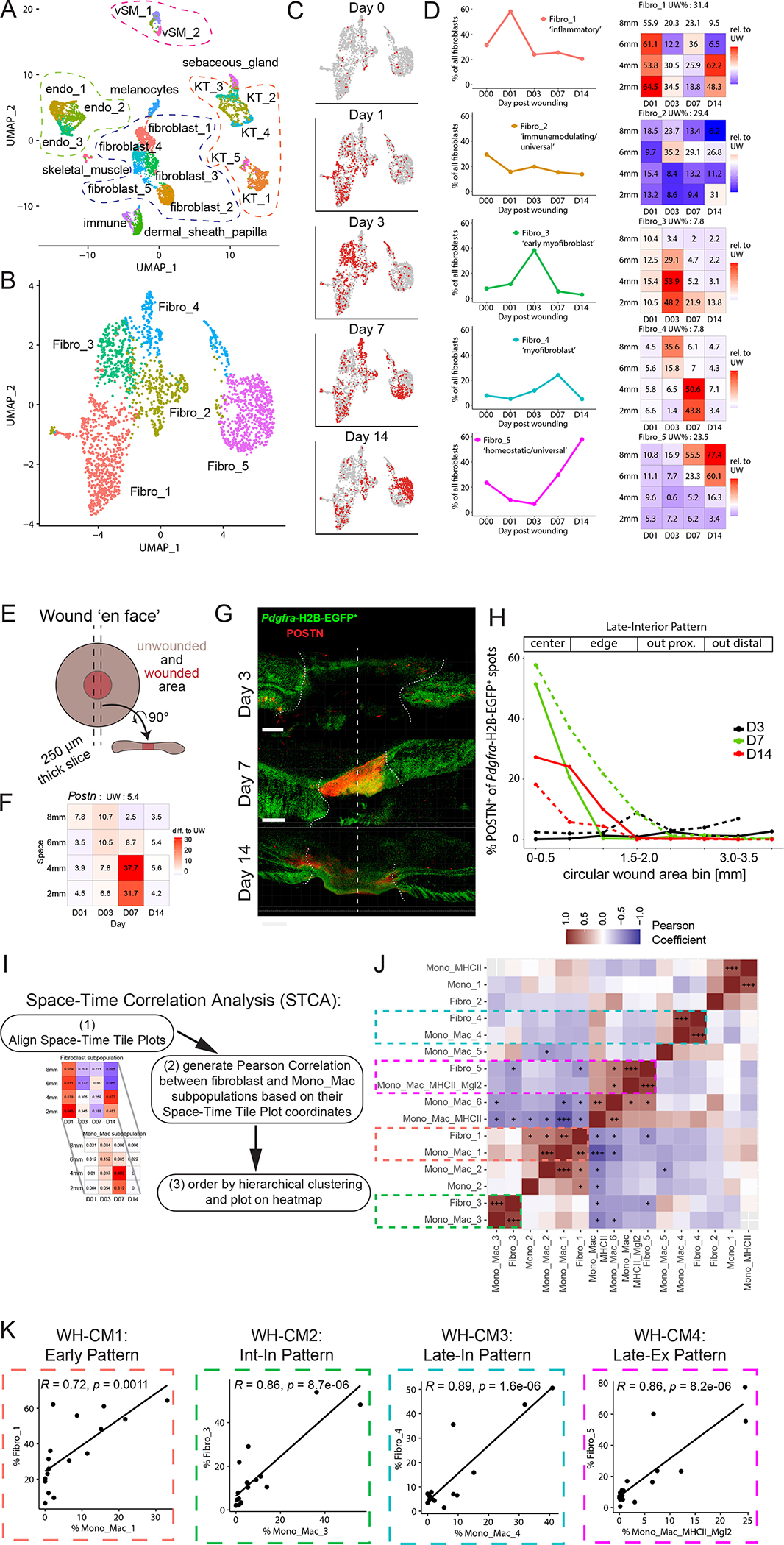
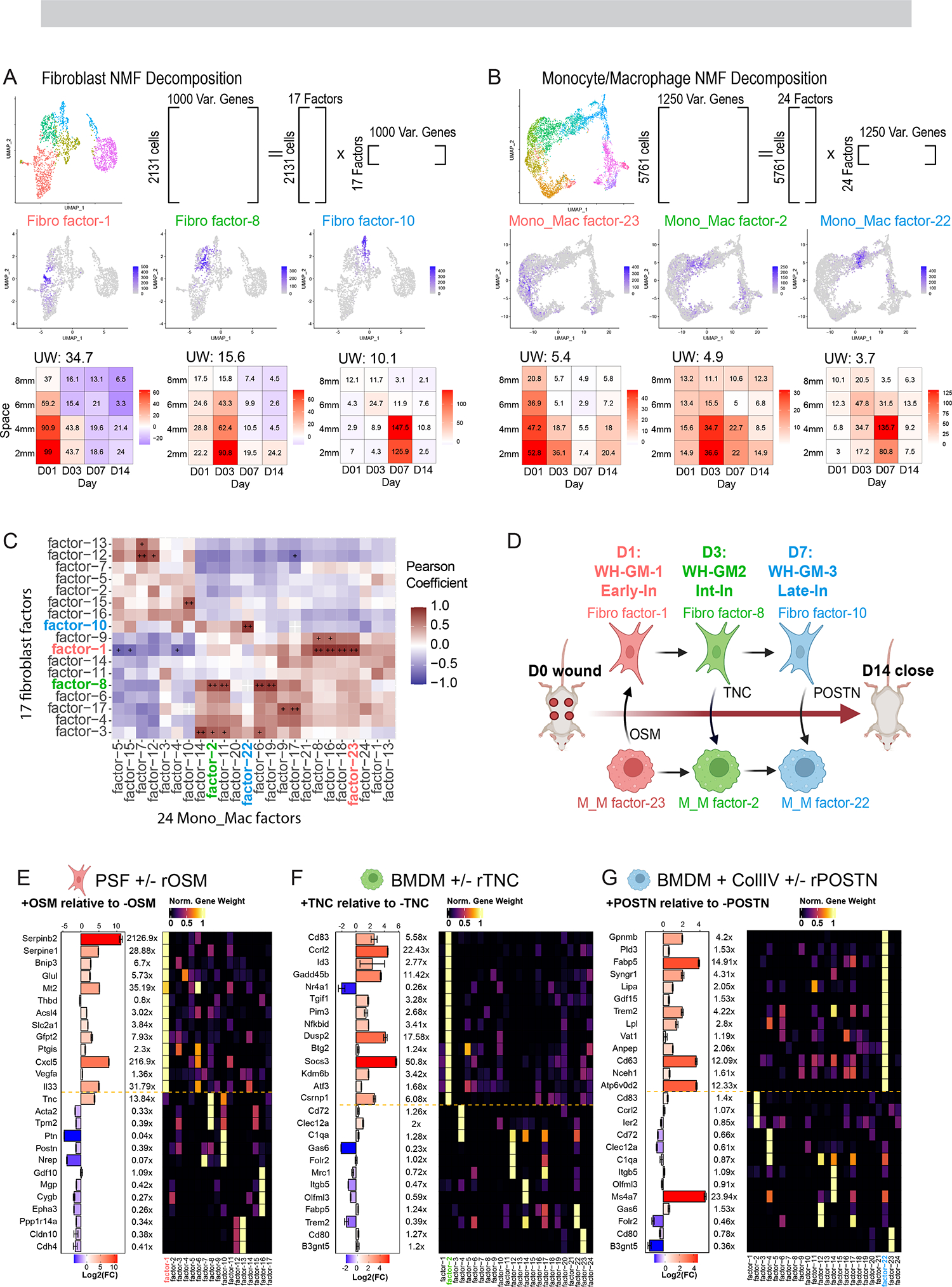
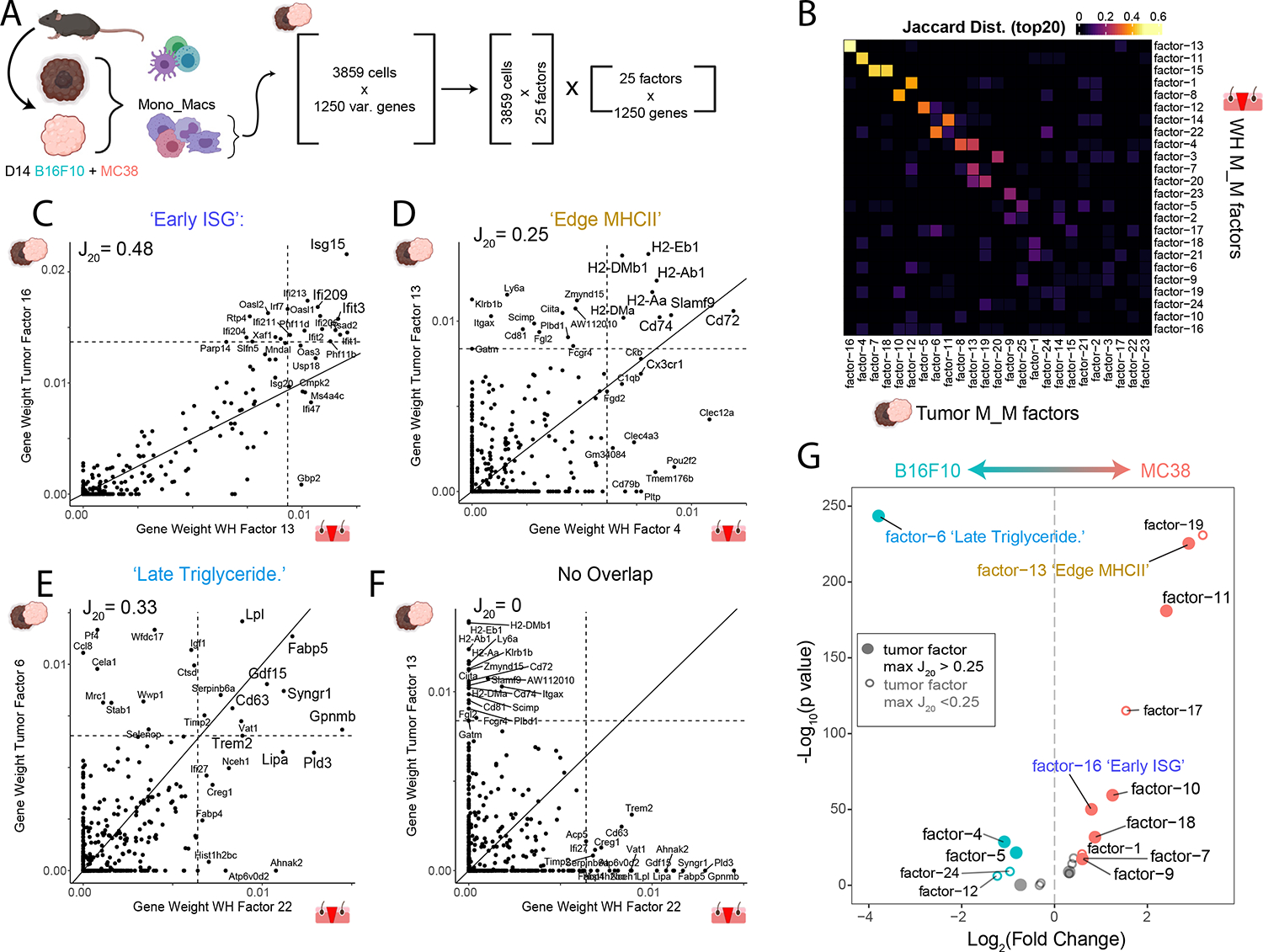
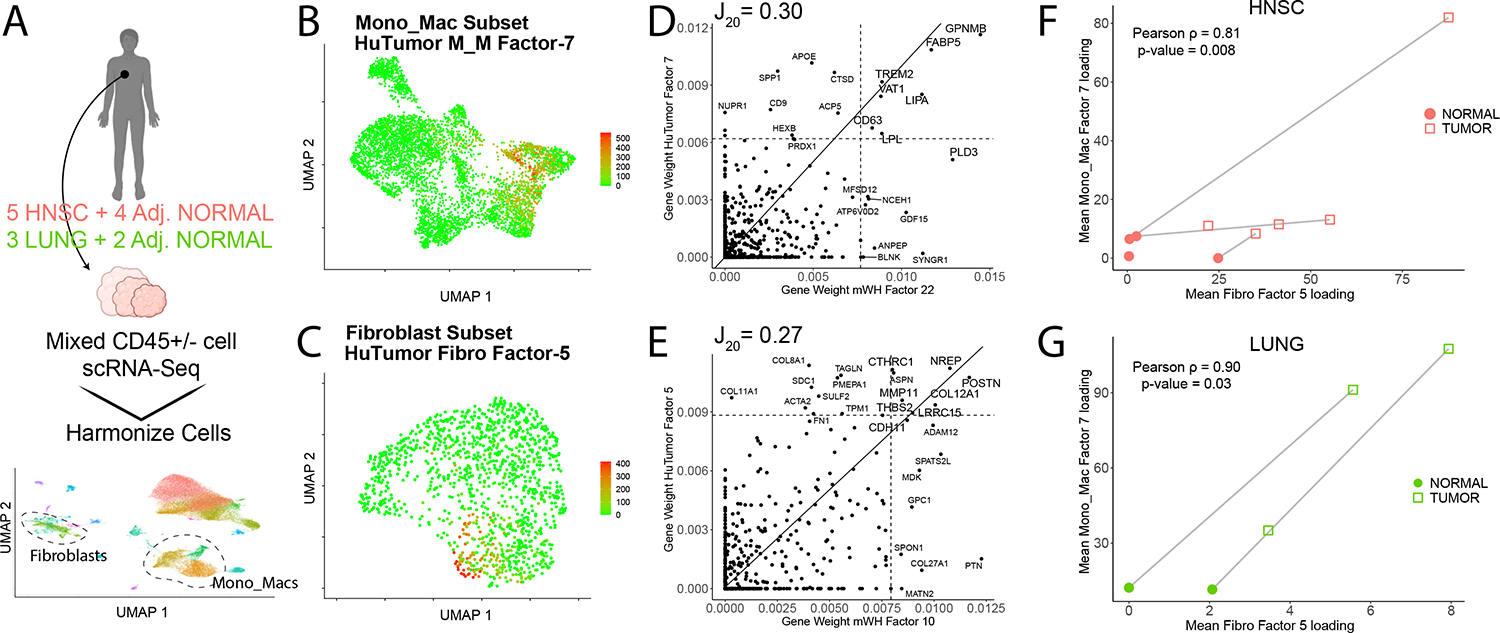
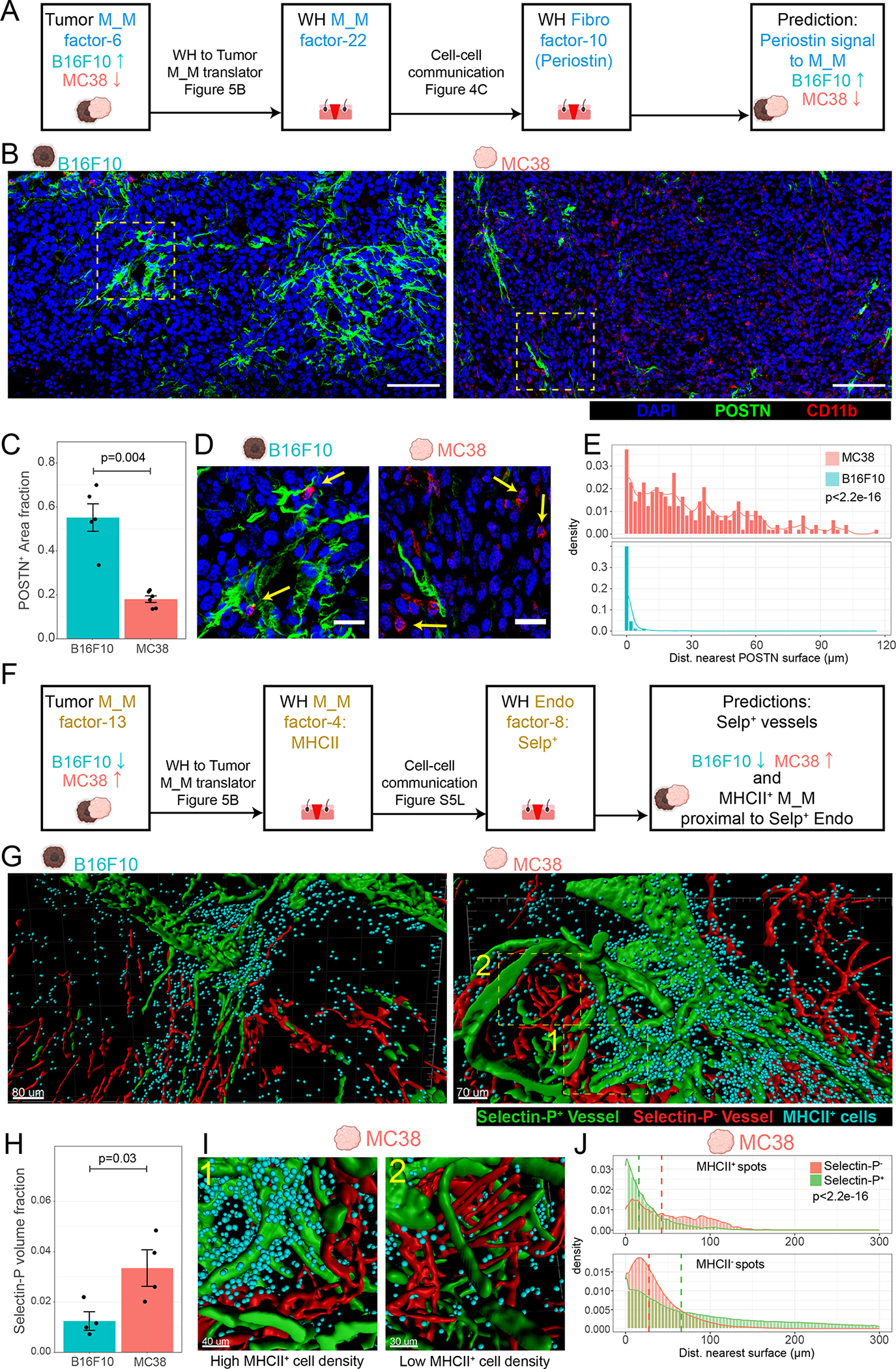
Tissue repair responses in metazoans are highly coordinated by different cell types over space and time. However, comprehensive single-cell-based characterization covering this coordination is lacking. Here, we captured transcriptional states of single cells over space and time during skin wound closure, revealing choreographed gene-expression profiles. We identified shared space-time patterns of cellular and gene program enrichment, which we call multicellular "movements" spanning multiple cell types. We validated some of the discovered space-time movements using large-volume imaging of cleared wounds and demonstrated the value of this analysis to predict "sender" and "receiver" gene programs in macrophages and fibroblasts. Finally, we tested the hypothesis that tumors are like "wounds that never heal" and found conserved wound healing movements in mouse melanoma and colorectal tumor models, as well as human tumor samples, revealing fundamental multicellular units of tissue biology for integrative studies.
Keywords: cell-cell crosstalk; fibroblasts; gene programs; immunology; macrophage; skin; systems biology; tumor microenvironment; wound healing.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.F.K. is a founder and shareholder of PIONYR immunotherapeutic and FOUNDERY innovations.
Figures
Comment in
-
Choreographing tissue repair.Nat Immunol. 2023 Jul;24(7):1051. doi: 10.1038/s41590-023-01556-4. Nat Immunol. 2023. PMID: 37340181 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
