

SHP2 Inhibition Sensitizes Diverse Oncogene-Addicted Solid Tumors to Re-treatment with Targeted Therapy
- PMID: 37269335
- PMCID: PMC10401072
- DOI: 10.1158/2159-8290.CD-23-0361
SHP2 Inhibition Sensitizes Diverse Oncogene-Addicted Solid Tumors to Re-treatment with Targeted Therapy
Abstract
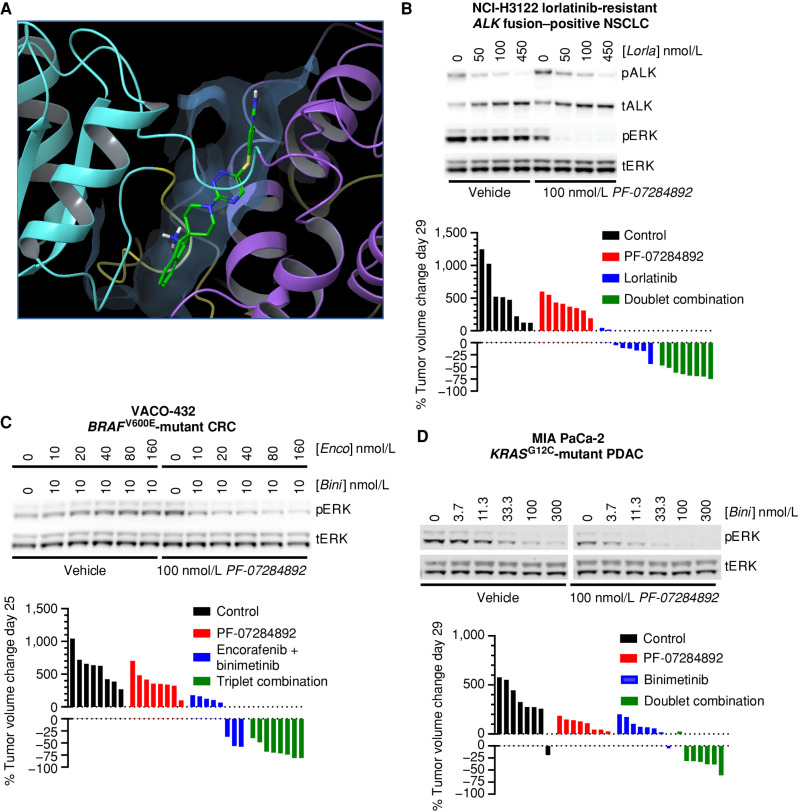
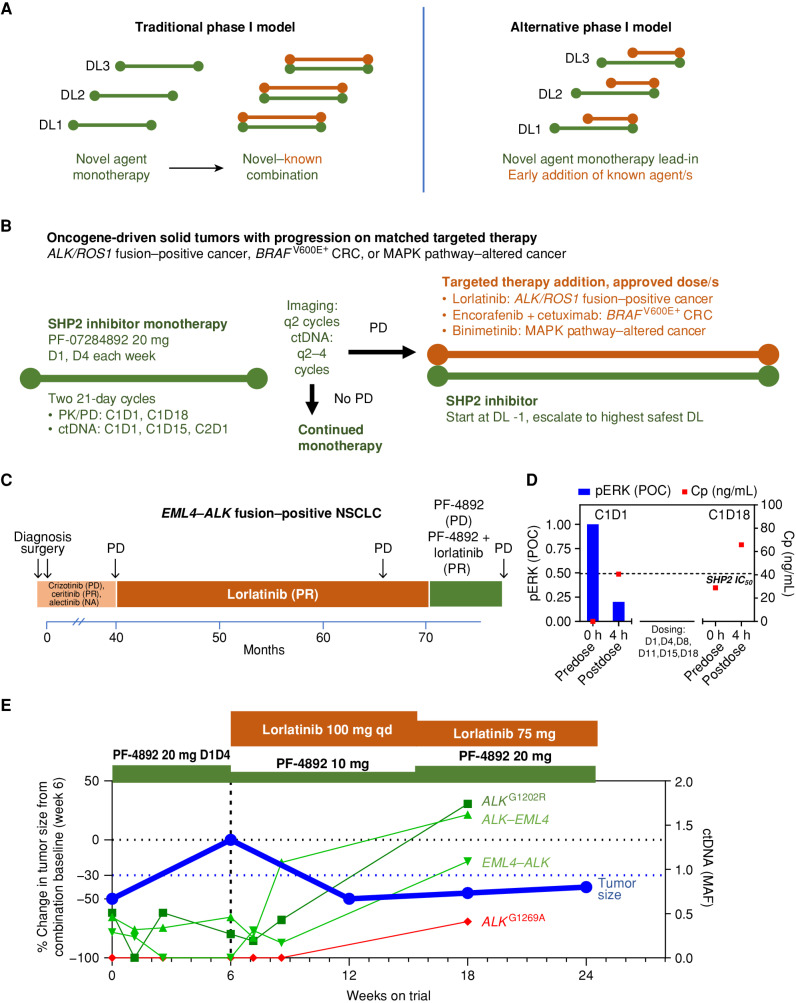
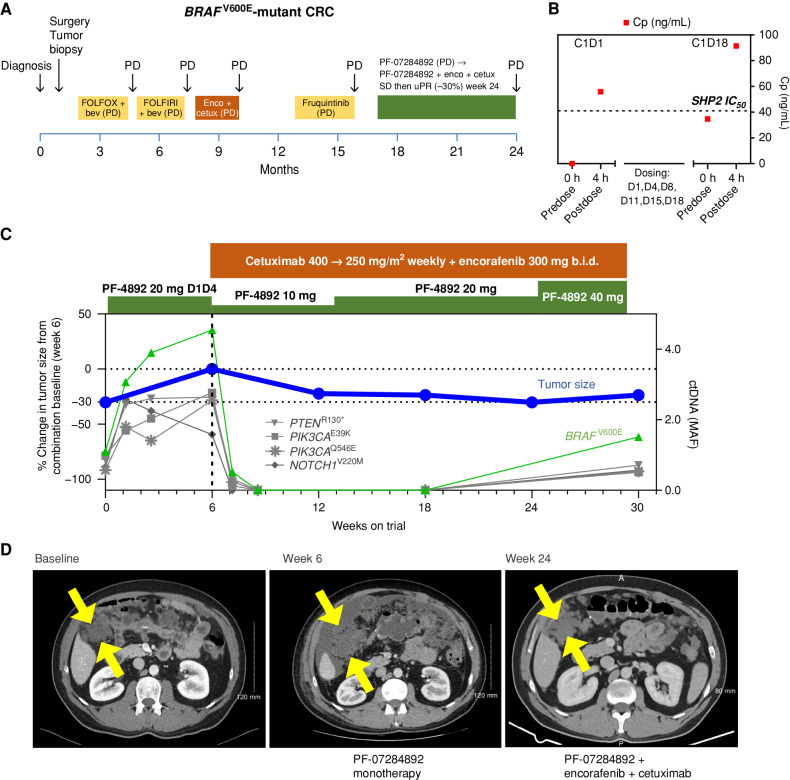
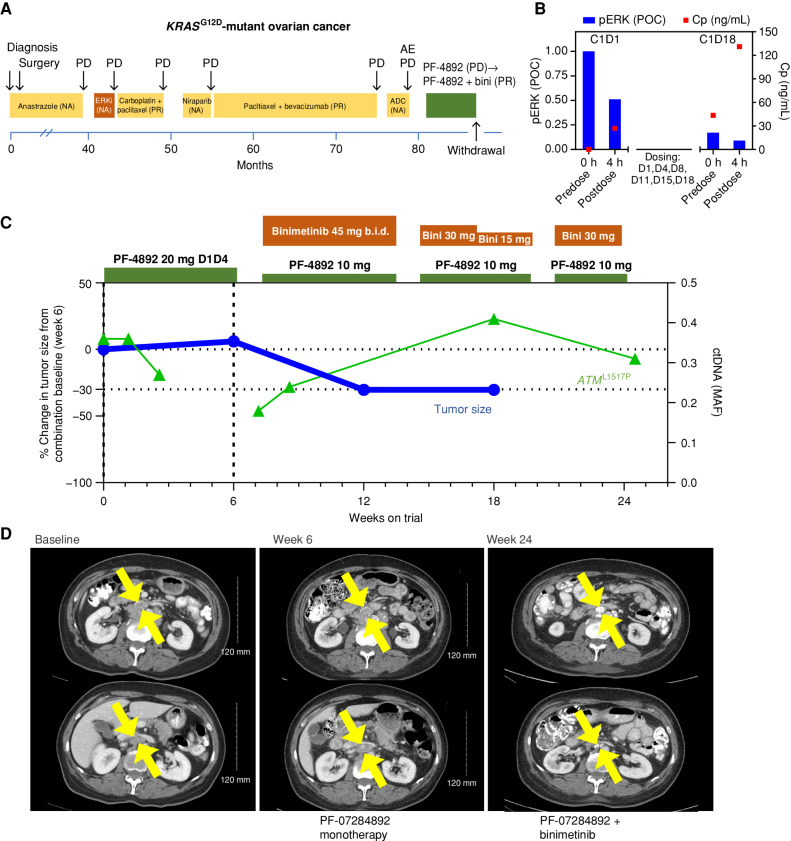
Rationally targeted therapies have transformed cancer treatment, but many patients develop resistance through bypass signaling pathway activation. PF-07284892 (ARRY-558) is an allosteric SHP2 inhibitor designed to overcome bypass-signaling-mediated resistance when combined with inhibitors of various oncogenic drivers. Activity in this setting was confirmed in diverse tumor models. Patients with ALK fusion-positive lung cancer, BRAFV600E-mutant colorectal cancer, KRASG12D-mutant ovarian cancer, and ROS1 fusion-positive pancreatic cancer who previously developed targeted therapy resistance were treated with PF-07284892 on the first dose level of a first-in-human clinical trial. After progression on PF-07284892 monotherapy, a novel study design allowed the addition of oncogene-directed targeted therapy that had previously failed. Combination therapy led to rapid tumor and circulating tumor DNA (ctDNA) responses and extended the duration of overall clinical benefit.
Significance: PF-07284892-targeted therapy combinations overcame bypass-signaling-mediated resistance in a clinical setting in which neither component was active on its own. This provides proof of concept of the utility of SHP2 inhibitors in overcoming resistance to diverse targeted therapies and provides a paradigm for accelerated testing of novel drug combinations early in clinical development. See related commentary by Hernando-Calvo and Garralda, p. 1762. This article is highlighted in the In This Issue feature, p. 1749.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
-
Patient-Centric Approaches for Phase I Combination Trials Come on Stage.Cancer Discov. 2023 Aug 4;13(8):1762-1764. doi: 10.1158/2159-8290.CD-23-0534. Cancer Discov. 2023. PMID: 37269291 Free PMC article.
References
-
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. . Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100–3. - PubMed
-
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. . MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
