

ITGA3 acts as a purity-independent biomarker of both immunotherapy and chemotherapy resistance in pancreatic cancer: bioinformatics and experimental analysis
- PMID: 37270717
- PMCID: PMC10239741
- DOI: 10.1007/s10142-023-01122-z
ITGA3 acts as a purity-independent biomarker of both immunotherapy and chemotherapy resistance in pancreatic cancer: bioinformatics and experimental analysis
Retraction in
-
Retraction Note: ITGA3 acts as a purity-independent biomarker of both immunotherapy and chemotherapy resistance in pancreatic cancer: bioinformatics and experimental analysis.Funct Integr Genomics. 2024 May 11;24(3):86. doi: 10.1007/s10142-024-01373-4. Funct Integr Genomics. 2024. PMID: 38730051 Free PMC article. No abstract available.
Abstract
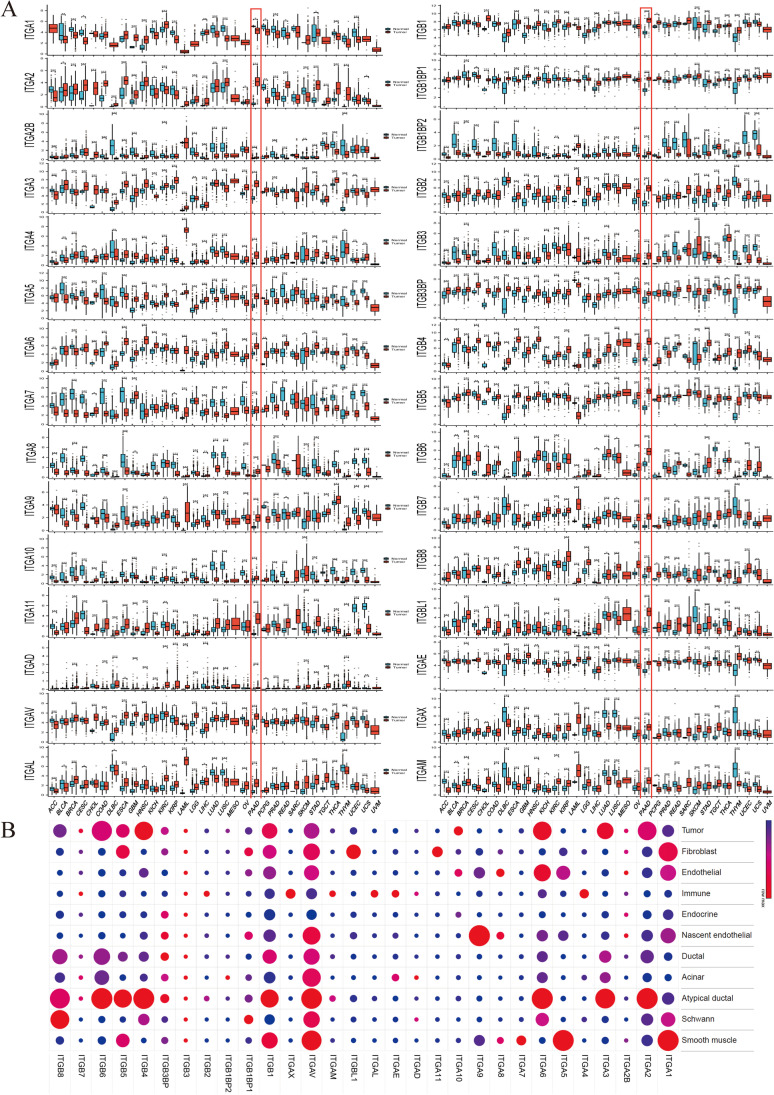
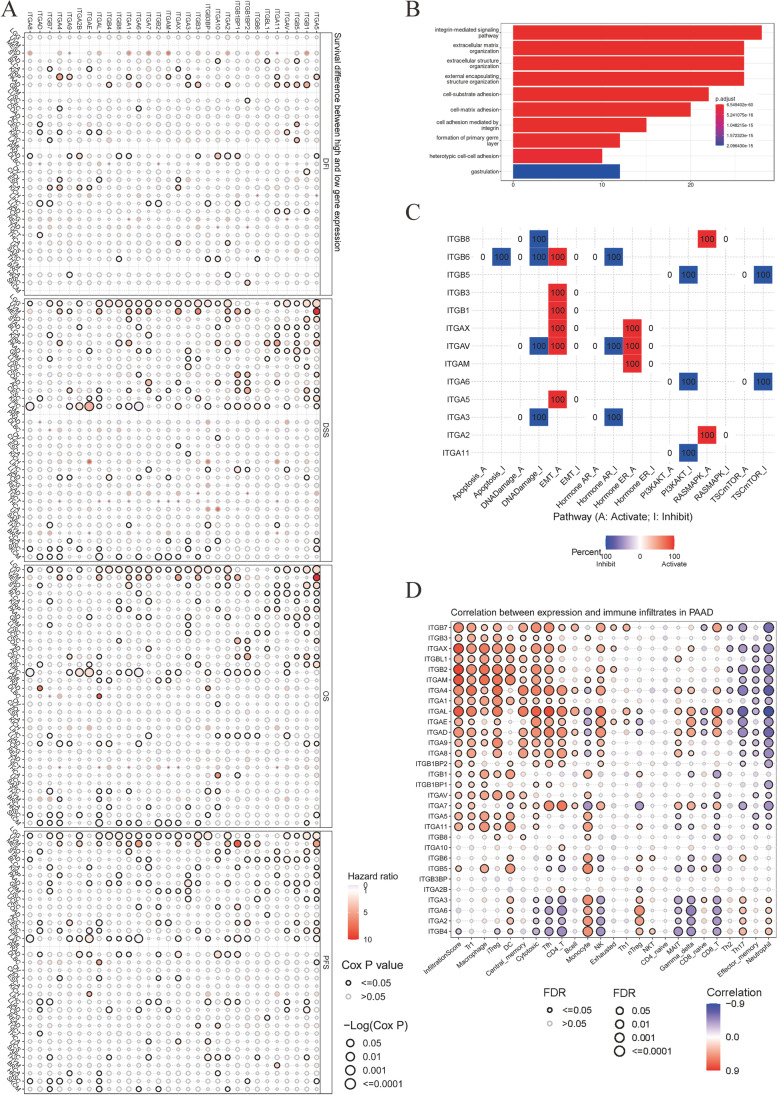
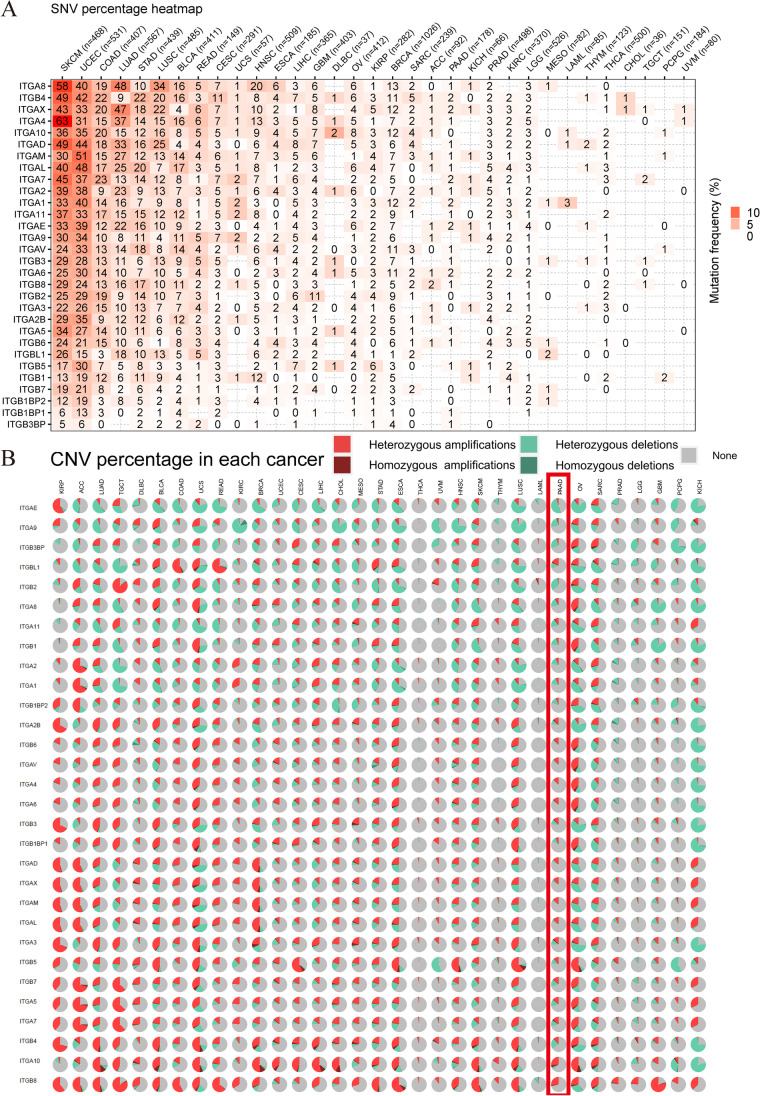
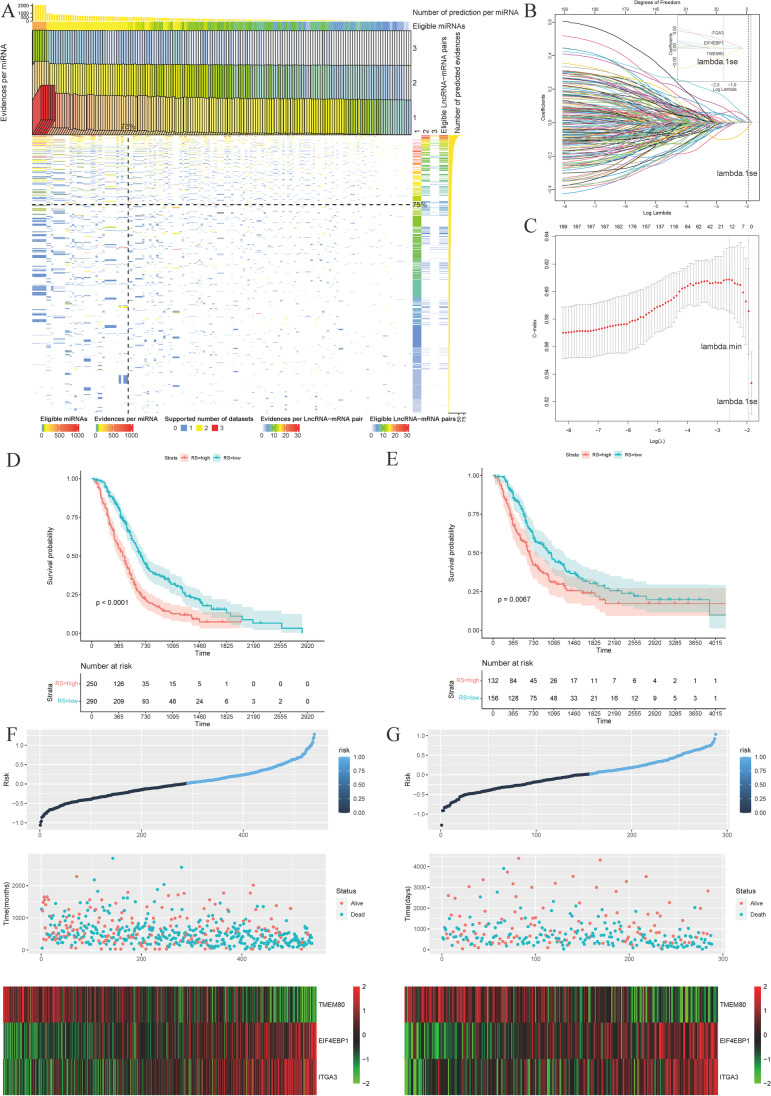
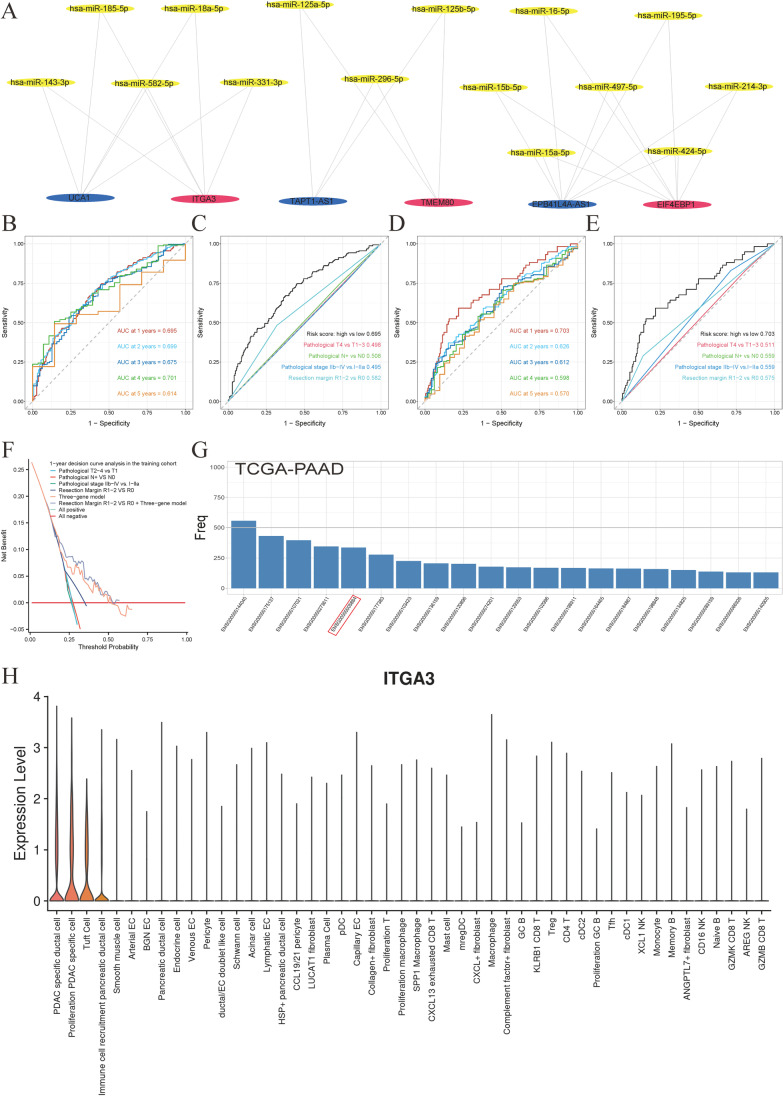
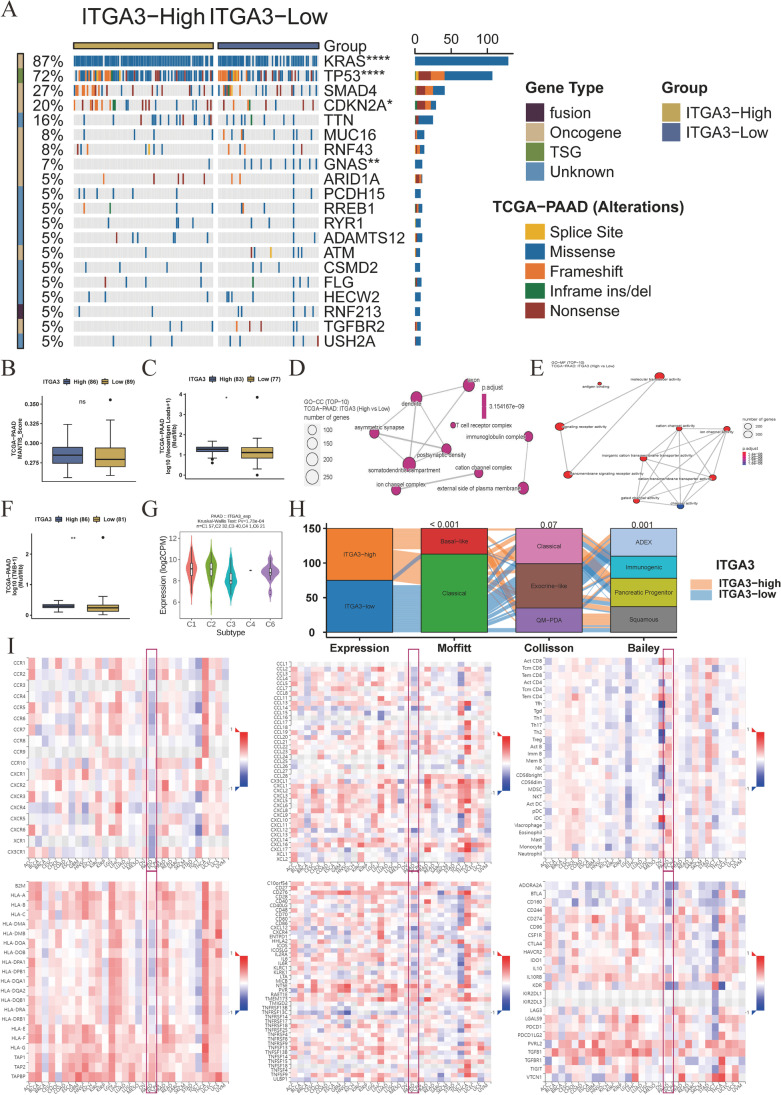
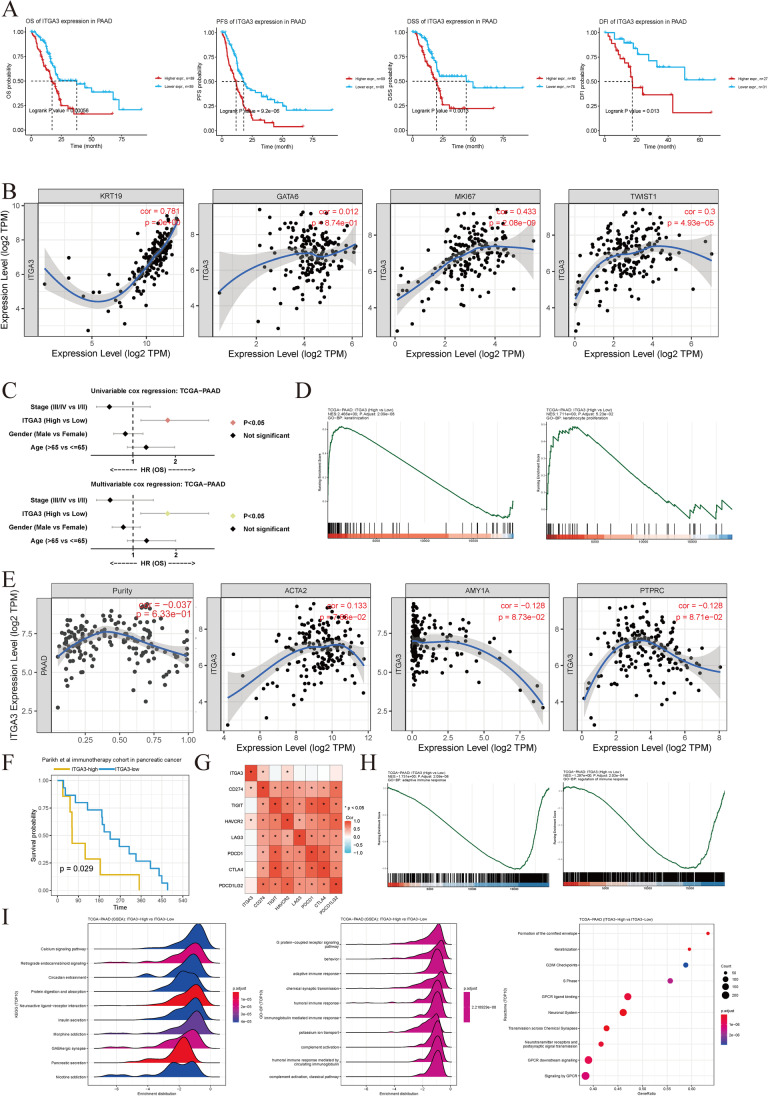
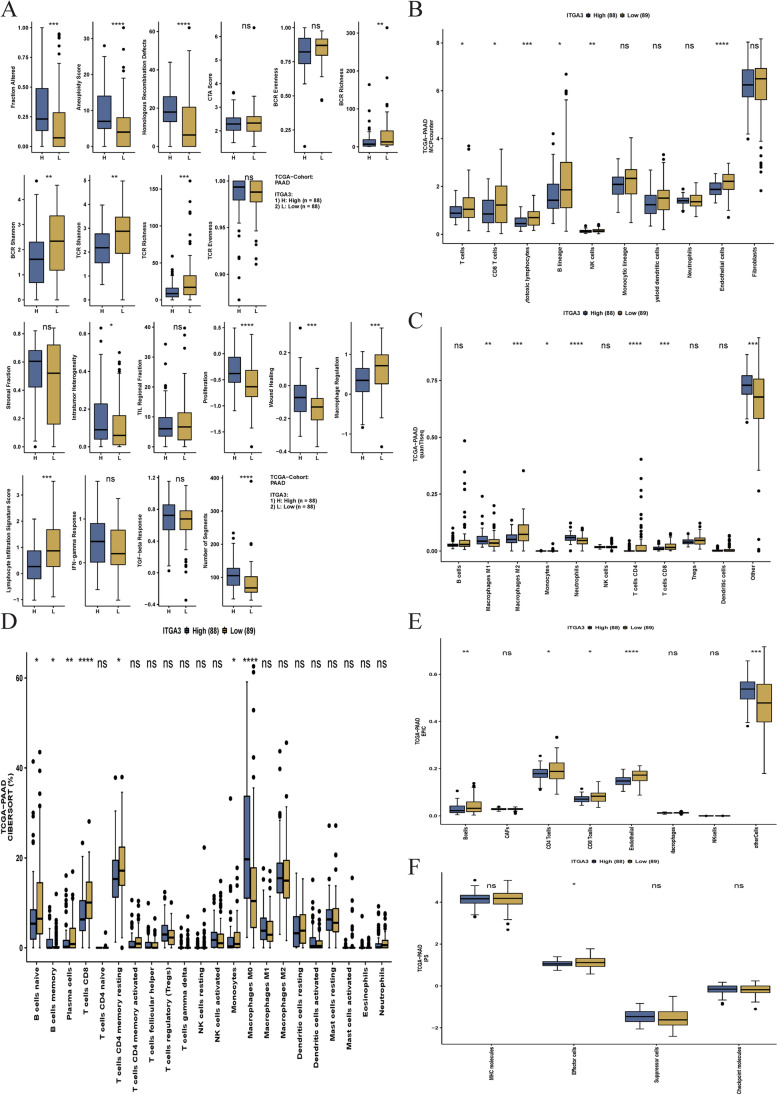
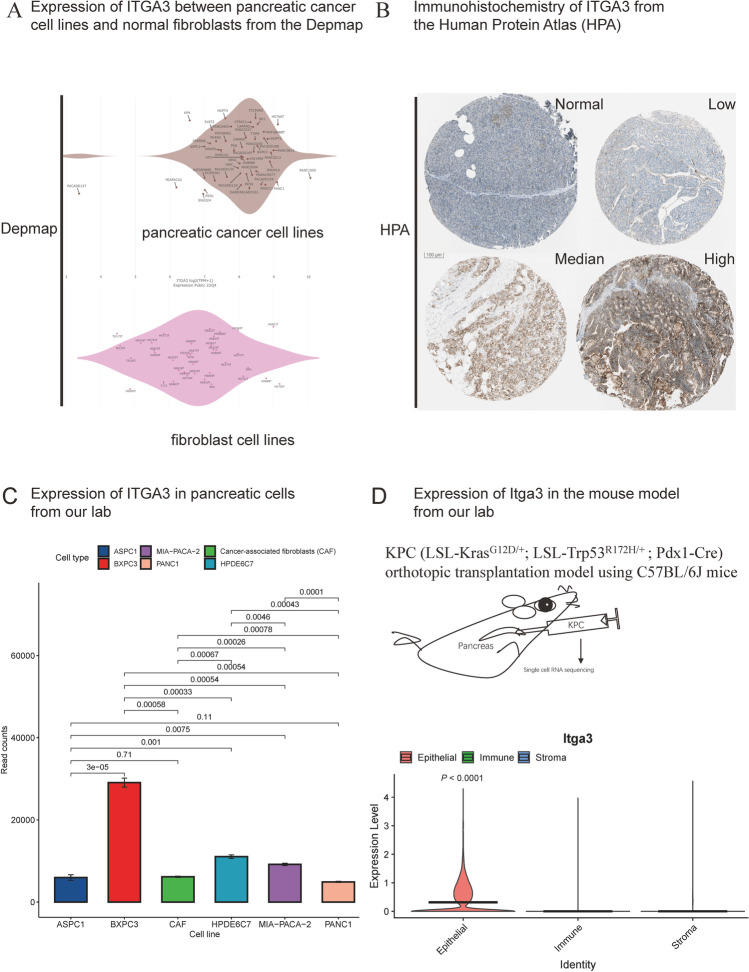
Contribution of integrin superfamily genes to treatment resistance remains uncertain. Genome patterns of thirty integrin superfamily genes were analyzed of using bulk and single-cell RNA sequencing, mutation, copy number, methylation, clinical information, immune cell infiltration, and drug sensitivity data. To select the integrins that are most strongly associated with treatment resistance in pancreatic cancer, a purity-independent RNA regulation network including integrins were constructed using machine learning. The integrin superfamily genes exhibit extensive dysregulated expression, genome alterations, epigenetic modifications, immune cell infiltration, and drug sensitivity, as evidenced by multi-omics data. However, their heterogeneity varies among different cancers. After constructing a three-gene (TMEM80, EIF4EBP1, and ITGA3) purity-independent Cox regression model using machine learning, ITGA3 was identified as a critical integrin subunit gene in pancreatic cancer. ITGA3 is involved in the molecular transformation from the classical to the basal subtype in pancreatic cancer. Elevated ITGA3 expression correlated with a malignant phenotype characterized by higher PD-L1 expression and reduced CD8+ T cell infiltration, resulting in unfavorable outcomes in patients receiving either chemotherapy or immunotherapy. Our findings suggest that ITGA3 is an important integrin in pancreatic cancer, contributing to chemotherapy resistance and immune checkpoint blockade therapy resistance.
Keywords: Chemotherapy resistance; ITGA3; Immunotherapy resistance; Integrin; Machine learning; Pancreatic cancer.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Akagbosu B, Tayyebi Z, Shibu G, Paucar Iza YA, Deep D, Parisotto YF, Fisher L, Pasolli HA, Thevin V, Elmentaite R, Knott M, Hemmers S, Jahn L, Friedrich C, Verter J, Wang ZM, van den Brink M, Gasteiger G, Grunewald TGP, Marie JC, Leslie C, Rudensky AY, Brown CC. Novel antigen-presenting cell imparts T (reg)-dependent tolerance to gut microbiota. Nature. 2022;610(7933):752–760. doi: 10.1038/s41586-022-05309-5. - DOI - PMC - PubMed
-
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–172. doi: 10.1038/s41590-018-0276-y. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
